
記住我
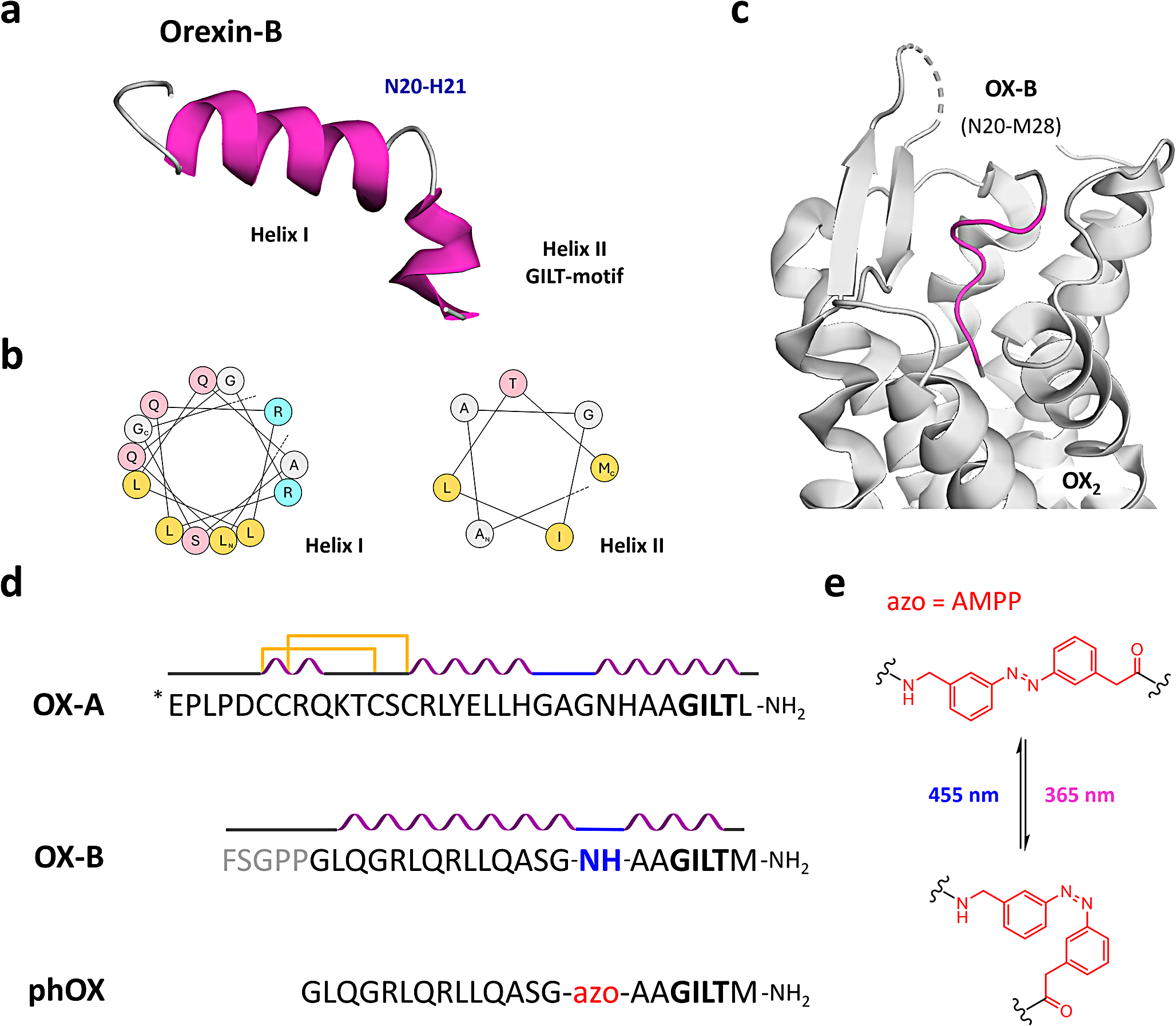

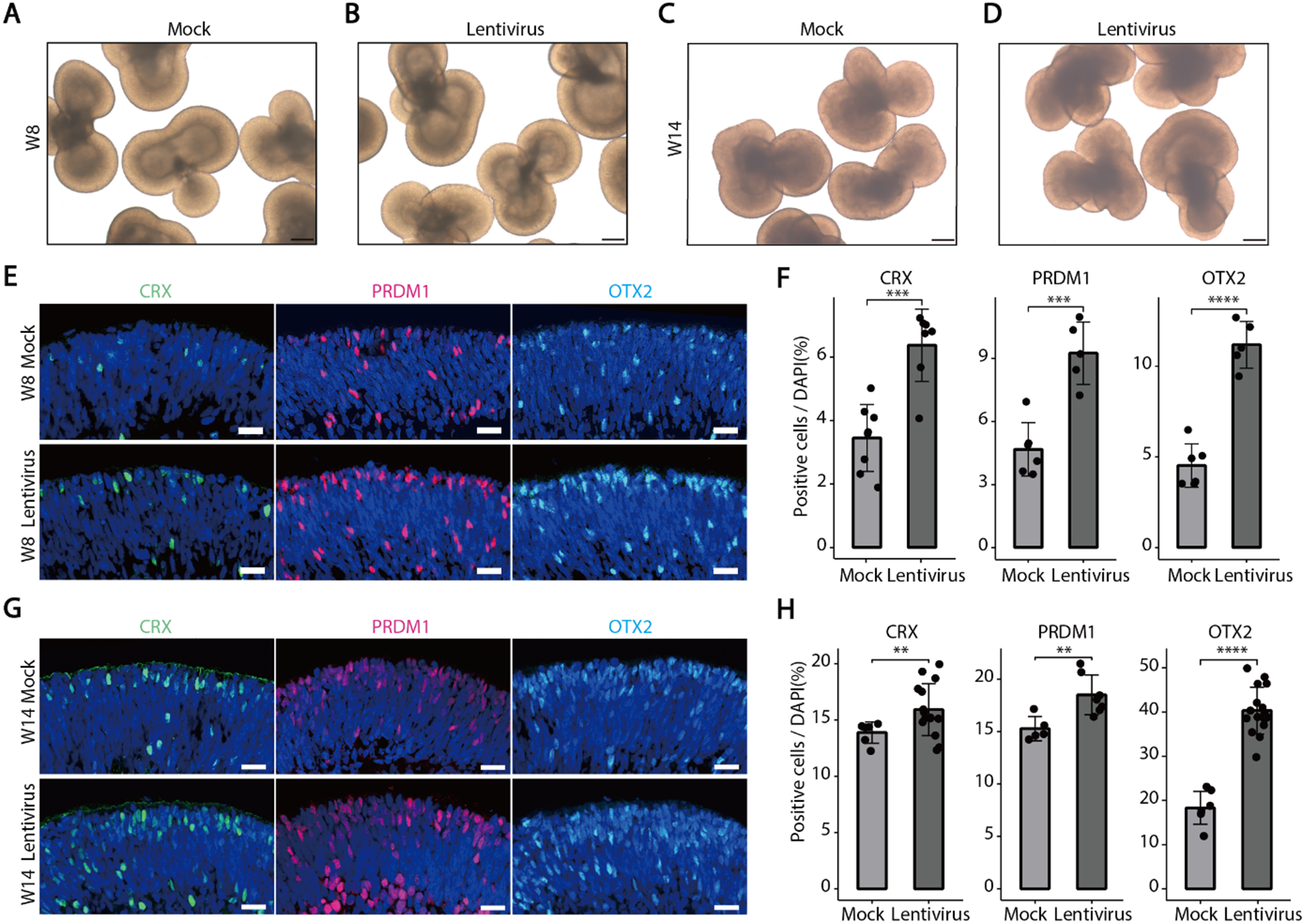

In this research, a precise and thoughtful arrangement of five unique experimental conditions was established, each centered on BJFF human organoids, collectively involving 118 replicates. Notably, the inception of the project took place on day 35, representing a significant juncture in their development. To evaluate the effect of the optimal freezing method, vitrification, on kidney organoids of various ages and to determine its applicability across different developmental stages, a trial was conducted using organoids at day 14 and day 21 (in addition to the primary organoids on day 35) [Fig. 8]. Vitrification was found to be effective across all ages tested. Day 35 kidney organoids were chosen in this study because organoid transporter activities become more matured at the late stage around day 42 ∼ 49 for cisplatin toxicity test. To test freezing methods, organoids were cryopreserved by 5 conditions structured as follows: one condition served as the control group, two conditions were designated for the slow freezing method, and the remaining two conditions were designated for vitrification [Fig. 9A].
Fig. 8
Illustration of the experiment testing the vitrification method on organoids of different ages. Additional experiment testing 5 μM cisplatin toxicity on organoids at 14, 21, and 35 days to demonstrate the effectiveness of vitrification across different ages. After thawing, organoids were cultured until day 49 of differentiation and exposed to cisplatin for 24 h. Samples were harvested on day 56 of differentiation for immunohistochemical assessment (LTL: red, CDH1: green, PODXL: white). For the day 35 kidney organoids, they were thawed from our stocks after 7 weeks to check the effect of storage in the − 150 °C freezer. Scale bars: 80 μm
Fig. 9
Illustrating the experimental conditions and methodologies utilized for different freezing techniques in this study. (A) This study incorporated five distinct experimental conditions, delineating the specifics of each component. All replicates originated from human stem cells, with the kidney organoids being of BJFF type. (B) In-depth delineation of slow freezing techniques. The upper section pertains to Slow Freezing number one (SF1), while the lower section pertains to Slow Freezing number two (SF2). All procedural steps remained identical, barring the variation in freezing solutions utilized in this experimental setup. Providing comprehensive elaboration on the sequential steps involved: A- Extracting four organoids using the pipette at its lowest power to minimize media volume. B- Transferring replicates into cryotubes containing 200 μl of freezing media; the upper cryotube (SF1) contains 10% DMSO and advanced RPMI with glutamax, while the lower one (SF2) contains commercial freezing media (Bambanker). C- Ensuring the proper closure of cryotube caps. D- Placing cryotubes in a -80 degree freezer for 24 h. E- Moving cryotubes to a -150-degree freezer for an additional 6 days. F - Retrieving cryotubes after 7 days of freezing for thawing. G - Adding 1 milliliter of fresh room-temperature media for thawing. H - Gently extract replicates using the pipette at the lowest possible power. I - Transferring them into new 96-well plates. J - Moving all 14 replicates of each condition onto the same plate. K - Placing plates into the incubator for regular growth. (C) Detailed explanation of ultra-rapid freezing (Vitrification) techniques. The upper segment denotes Vitrification number one (V1), while the lower segment pertains to Vitrification number two (V2). All steps within the process remained consistent, differing solely in the equilibration, vitrification, and warming solutions applied in this experimental setup. A-Extracting four organoids using the pipette at minimal power to minimize media volume. B-Transferring replicates into Eppendorf tubes containing 200 μl of equilibration solution. The upper tube (ES1) comprises 10% DMSO, 10% Ethylene glycol in advanced RPMI with glutamax, and the lower tube (ES2) holds 7.5% DMSO, 7.5% Ethylene glycol in advanced RPMI with glutamax. C-Immerse the organoids into an equilibration solution at room temperature, with exposure times averaging 8–15 min to allow penetration of cryopreserved agents into the kidney organoids. D-Removing replicates to transfer them into specific vitrification solutions. E- Placing replicates into the vitrification solutions; each containing 200 μl. Vitrification solution 1 (VS1) comprises 20% DMSO, 20% Ethylene glycol in advanced RPMI with glutamax, while Vitrification solution 2 (VS2) includes 15% DMSO, 15% Ethylene glycol in advanced RPMI with glutamax. Exposure time should not exceed 30 s due to high toxicity; this process took less than 10 s in this experiment. F- Directly exposing closed-cap cryotubes to liquid nitrogen for 10 s. G- Placing cryotubes directly into the − 150-degree freezer for 7 days. H- Retrieving cryotubes after 7 days of freezing. I- Adding 1 milliliter of fresh room-temperature RPMI media for thawing. J- Removing replicates for multi-step warming solutions. K- Placing organoids into specific warming solutions; each containing 200 μl. *- V1 warming involves 3 steps: 0.5 M sucrose in advanced RPMI with glutamax for 3 min, followed by 0.25 M sucrose for 2 min, and lastly, 0.125 M sucrose for 2 min. **- V2 warming entails 2 steps: 0.3 M sucrose for 3 min then 0.2 M sucrose for 2 min in advanced RPMI with glutamax. L- Retrieving replicates for placement into new 96-well plates. M- Transferring them into the new 96-well plate. N- Moving all 14 organoids of each condition into the same plate. O- Placing the plates into the incubator for regular growth
Control groupAmong these five conditions, one functioned as the control group and was exposed to conventional incubator culture conditions. In this particular group, a well-considered approach to media replacement was implemented. Every two days, the media in each replicate was renewed, with each replacement amounting to half of the total media volume. This systematic procedure was employed to guarantee the preservation of an ideal culture environment.
Slow-freezing conditionsTwo conditions were designated for slow-freezing processes [Fig. 9B], labeled as SF1 and SF2. In SF1, the organoids encountered a freezing solution composed of 10% Dimethyl Sulfoxide (DMSO) in advanced Roswell Park Memorial Institute (RPMI) culture media, enriched with Glutamax. Meanwhile, SF2 involved the use of a commercial freezing medium known as “Bambanker”.
In both slow-freezing conditions, 200 μl of freezing medium were loaded into each cryotube, with two replicates for redundancy. The slow-freezing process commenced with an initial storage phase in a -80 °C freezer, followed by a transfer to a -150 °C freezer, resulting in a total freezing period of seven days. Subsequently, for both slow-freezing conditions, the thawing procedure consisted of adding one milliliter of fresh media at room temperature to minimize any dilution of the freezing medium, followed by gentle pipetting steps.
Following this, a comprehensive washing process was conducted using an extra milliliter of RPMI at room temperature. Subsequently, the organoids were retrieved using wide bore tips, reducing the media volume, and then re-suspended in 96-well plates. Each well contained 200 μl of fresh RPMI, pre-warmed and enriched with Glutamax.
Similar to the control group, media replenishment in these conditions involved the replacement of half of the media every two days. Following a seven-day period, all the replicates from these conditions were subjected to a series of tests to evaluate their responses and viability.
Vitrification conditionsTwo more experimental conditions were specifically assigned to the application of vitrification techniques [Fig. 9C]. These conditions were differentiated by the concentrations of cryoprotectants (CPAs) employed. To demonstrate these conditions, we employed two well-established methods commonly used for cryopreservation of tissues and cells, particularly in the field of gynecology (specifically for human blastocytes), which are widely recognized and utilized [27, 28]. In each vitrification condition, a series of three stages was meticulously carried out, encompassing equilibration (aimed at enabling CPA penetration into the organoid structures), vitrification, and subsequent warming solutions.
Following a specified duration in the equilibration solution, the organoids were moved to the vitrification solution, which featured higher CPA concentrations. After this step, the organoids were rapidly immersed in liquid nitrogen for a very brief duration, not exceeding 30 s, to ensure vitrification. Thawing procedures differed between the two vitrification conditions and involved the use of varying sucrose dilutions. The organoids were then placed in 96-well plates containing pre-warmed advanced RPMI culture media with Glutamax, making them ready for further use.
In the initial vitrification condition, denoted as V1 or Vitrification number 1, the equilibration solution consisted of a combination of 10% DMSO and 10% Ethylene Glycol, as per the protocol adapted from the study by Michelle Lane et al. [27]. The organoids were subjected to this equilibration solution for a duration ranging from 8 to 15 min at room temperature, and exposure time varies based on the replicate size.
After the equilibration phase, all replicates were expeditiously moved into cryotubes filled with 200 μl of vitrification solution. This solution consisted of 20% DMSO, 20% Ethylene Glycol, and 0.75 M sucrose. It’s important to highlight that the transfer to the vitrification solution and the subsequent steps for cryopreservation were completed swiftly, taking no more than 30 s, with most lasting less than 10 s.
The cryotubes were promptly submerged into liquid nitrogen to commence the vitrification procedure, followed by placement in a -150 °C freezer. It’s noteworthy that each cryotube contained two replicates, allowing for a thorough evaluation.
After a meticulously defined 7-day vitrification period, the thawing process was carried out with precision. It involved the use of 1 milliliter of fresh media at room temperature, primarily aimed at decreasing the ice blocker concentration. Following this, a three-step warming technique was applied to ensure the gradual recovery of the organoids.
At the outset, the organoids were moved to microtubes filled with 200-microliter volumes of 0.5 M sucrose solution in advanced RPMI for a 3-minute interval. Subsequently, they were relocated to another tube with 200 μl of 0.25 M sucrose solution in advanced RPMI, undergoing an equivalent 3-minute duration. Finally, the organoids were introduced to a tube containing 200 μl of 0.125 M sucrose solution for 2 min, effectively completing the gradual warming process.
After these thorough processes, the organoids were relocated to 96-well plates, and, in adherence to the prescribed protocol, half of the culture media was renewed every two days, guaranteeing their continued viability and growth during the post-thaw culture phase.
In the second vitrification condition, labeled as V2 or Vitrification number 2, the equilibration solution consisted of 7.5% DMSO and 7.5% Ethylene Glycol, following the protocol derived from the research conducted by Tetsunori Mukaida et al. [28]. The organoids were immersed in this equilibration solution for a specific period, which varied based on the size of the replicates and typically ranged from 8 to 15 min at room temperature. Following the equilibration step, the organoids were transferred to individual cryotubes, each containing 200 μl of the vitrification solution.
The vitrification solution in this case contained a mixture of 15% DMSO and 15% Ethylene Glycol, both suspended in advanced RPMI with Glutamax. Similar to the procedure in the first vitrification condition, the transition from equilibration to vitrification was executed rapidly, taking no more than 30 s (with an actual duration of less than 10 s). Subsequently, the cryotubes, each accommodating the organoid replicates, were immediately submerged in liquid nitrogen, marking the initiation of the vitrification process. Following this step, the cryotubes were transferred to a -150 °C freezer and kept there for a precisely defined 7-day duration.
After the completion of the 7-day vitrification period, the thawing process was carried out meticulously. To initiate thawing and reduce the concentration of ice blockers, 1 milliliter of fresh RPMI at room temperature was utilized. The subsequent warming process involved a two-step method. Firstly, the organoids were transferred to microtubes containing 200 μl of a 0.3 M sucrose solution dissolved in fresh RPMI at room temperature, and this step was maintained for 3 min.
Following this, they were transferred to another tube containing 200 μl of a 0.2 M sucrose solution in fresh RPMI at room temperature for an equivalent duration of 2 min. To conclude the stepwise warming process, the organoids were placed in 96-well plates, with each well containing 200 μl of fresh RPMI pre-warmed to room temperature. Just like in all the other conditions, the standard protocol entailed the regular replacement of half the media every two days, ensuring the continual well-being and growth of the organoids during the post-thaw culture period.
Cell viability evaluation of the organoids after thawingUpon the conclusion of a 7-day incubation period (when the organoids had reached an age of 42 days), four replicates were chosen from each experimental condition. This was followed by a delicate washing step involving the use of 500 μl of phosphate-buffered saline (PBS) to remove the media. Subsequently, the organoids were subjected to 300 μl of a 0.25% Trypsin-Ethylenediaminetetraacetic acid (EDTA) solution and housed within incubators for a total duration of 15 min, with an initial 7-minute period followed by an additional 8 min.
During the initial 7-minute period, gentle pipetting was executed to ensure the effective exposure of the organoids to the Trypsin-EDTA solution. At the conclusion of the 15-minute incubation, cold RPMI was introduced to neutralize the Trypsin-EDTA, and additional gentle pipetting was carried out to aid the separation of the organoids into individual cells. The subsequent step entailed the use of a centrifuge machine for 4 min at a force of 300 g. Following centrifugation, the supernatant was precisely withdrawn, and 100 μl of RPMI, pre-warmed, were introduced to the cell pellet.
Thorough mixing was performed to ensure uniformity within the solution. To evaluate cell viability, 10 μl of the solution were mixed with an equal volume of trypan blue, and the quantification of cells was subsequently carried out using a specialized cell counting device.
Following the thawing process for all experimental conditions (excluding the control group), a comprehensive series of assessments was undertaken to examine the structural and functional aspects of the kidney organoids. To establish a benchmark, microscopic examinations were conducted by capturing images of the organoids before the initiation of the testing phase, thereby acquiring data concerning their initial dimensions. Subsequently, a decision was made to standardize the measurements for the sake of convenient comparisons. From the project’s inception on Day 0 (or in the case of freezing conditions, post-thawing), until the culmination of the experiment on Day 7, the dimensions of the replicates were subjected to daily scrutiny employing a light microscope and the Image J software. Upon standardization, meticulous comparisons were drawn across the different experimental conditions.
Quantitative reverse transcription PCR (qRT-PCR)Total RNA was extracted from kidney organoid samples utilizing TRIzol (Invitrogen) followed by assessment of concentration and purity using a NanoDrop (Thermo Fisher Scientific). Subsequently, cDNA synthesis was conducted from 400 ng of total RNA employing the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) as per the manufacturer’s instructions. Quantitative PCR was performed utilizing iTaq SYBR Green Supermix (Bio-Rad) on the QuantStudio3 Real-time PCR system. Relative gene expression levels were determined using the delta-delta CT method, with GAPDH serving as the housekeeping gene. Primer sequences utilized are provided in the Supplemental Table 1.
留言 (0)