
記住我

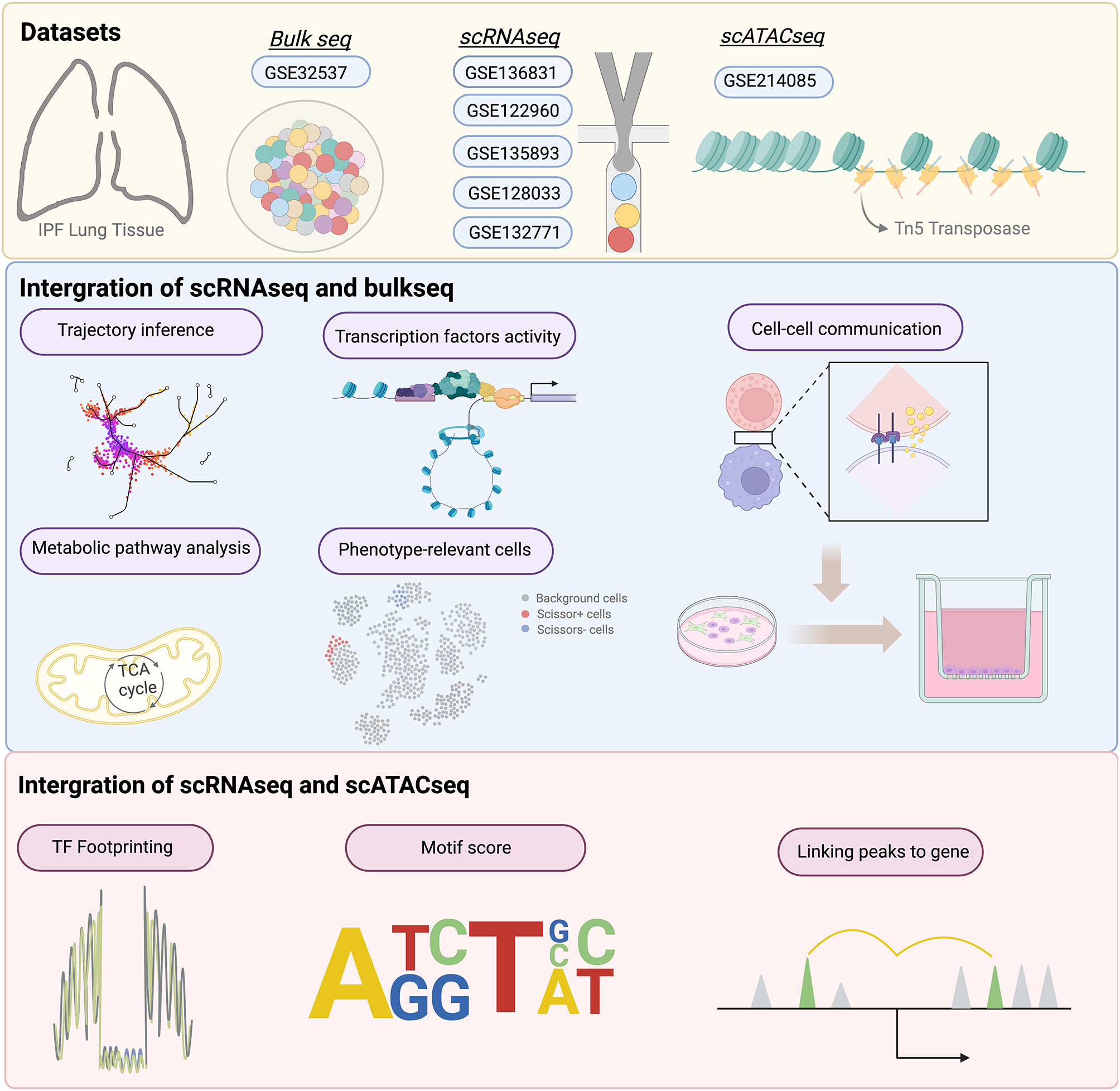
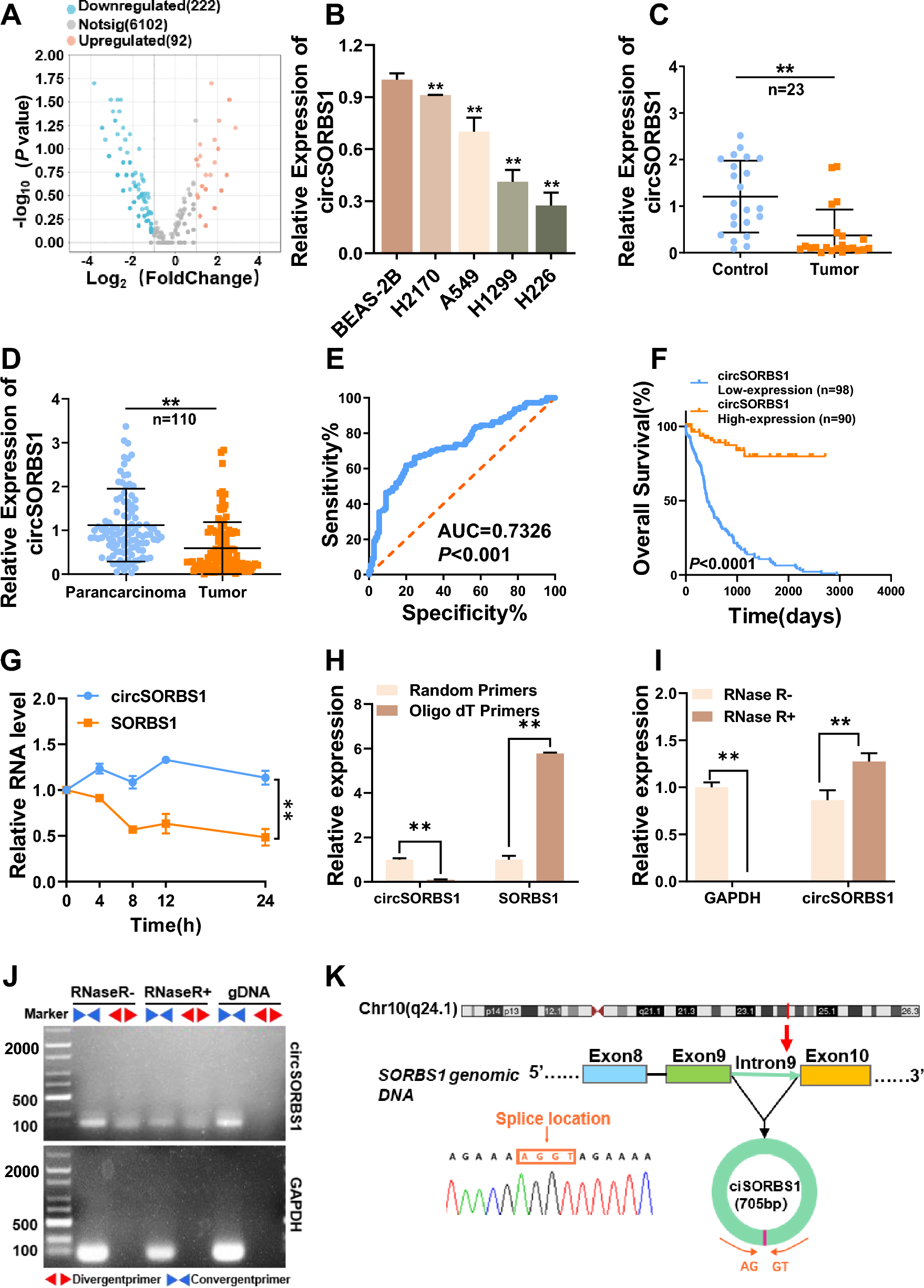


Figure 1 illustrates the analytical framework used in this study. RNA-seq data were obtained from the TCGA database for 575 CRC samples with more than 1 month of follow-up, and 51 normal colorectal tissues were used for comparison. Differential expression analysis was performed, and 8168 DEGs were identified in CRC when the threshold was set at |fold change|> 2 and FDR < 0.05. Of these genes, 4496 were upregulated and 3672 were downregulated (Fig. 2A). Additionally, 326 HRGs and 288 LMRGs were retrieved from the MSigDB database. We then conducted univariate Cox regression analysis on all protein-coding genes in the RNA-seq data of patients and identified 2207 genes associated with overall survival (OS). After taking the intersection of DEGs, genes associated with OS, HRGs, and LMRGs, we identified 26 hypoxia-related DEGs (HRDEGs) and 9 lactate metabolism-related DEGs (LMRDEGs, Fig. 2B, C). Analysis of these 35 genes revealed a wide range of correlations and intergene interactions (Fig. 2D, E).
Fig. 1
Outline of the analyses performed in this study. CRC: colorectal cancer; TCGA: The Cancer Genome Atlas; K–M: Kaplan–Meier; GSEA: gene set enrichment analysis; CMS: consensus molecular subtype; KEGG: Kyoto Encyclopedia of Genes and Genomes; GO: Gene Ontology; LASSO: least absolute shrinkage and selection operator; ROC: receiver operating characteristic; scRNA-seq, single-cell RNA-seq
Fig. 2
Identification of DEHRGs and DELMRGs in CRC. A Volcano plot of the DEGs between CRC and normal tissues. B Venn diagram of the HRGs, DEGs, and genes related to OS. C Venn diagram of the LMRGs, DEGs, and genes related to OS. D Heatmap of the correlations between the 35 DEHRGs and DELMRGs. E Interactions and gene function of the 35 DEHRGs and DELMRGs. F Heatmap for different numbers of clusters after unsupervised clustering. G Within-group clustering consistency at different numbers of clusters. DEHRGs: different expressed hypoxia-related genes; DELMRGs: different expressed lactate metabolism-related genes; CRC: colorectal cancer; OS: overall survival
Constructing molecular subtypes based on the HRDEGs and LMRDEGsUnsupervised clustering was performed on 575 CRC samples using the 35 identified HRDEGs and LMRDEGs. The highest average within-group agreement was observed when classified into three subtypes (Fig. 2F, G). Therefore, we classified the 575 samples into three subtypes: C1 (n = 92), C2 (n = 324), and C3 (n = 159). A comprehensive evaluation revealed that different molecular subtypes exhibited distinct hypoxia and lactate metabolism-related microenvironments (Figure S1A, B). The Kaplan–Meier survival curves indicated that patients with the C1 subtype had the worst OS than patients with subtypes C2 (HR = 1.67, 95% CI 1.07–2.62, p = 0.02) and C3 (HR = 2.48, 95% CI 1.41–4.36, p < 0.01). Additionally, there was a trend towards better OS in the C3 subtype compared with C2, although the difference was not statistically significant (C2 vs. C3, HR = 1.48, 95% CI 0.92–2.40, p = 0.11, Fig. 3A). The heat map in Fig. 3B shows the expression of the 35 genes in different subtypes and their association with clinical features. In particular, subtype C1 was associated with a higher number of patients with mucinous adenocarcinoma (MAC, p < 0.01) and advanced TNM stages (p < 0.01, Table 1). Additionally, we assessed the correlation between the molecular subtypes related to hypoxia and lactate metabolism and recognized CMS. Our findings revealed that the C1 subtype had a higher proportion of CMS4 (68.48%), whereas CMS2 dominated the C3 subtype (61.01%, Fig. 3C). GSEA of subtypes C1 and C3 revealed that the hypoxia pathway was significantly upregulated in C1 (FDR = 0.02). In contrast, the oxidative phosphorylation pathway was significantly downregulated (FDR = 0.01, Fig. 3D), consistent with our expectations. In addition, C1 exhibited a significant upregulation of KRAS signaling, EMT, and angiogenesis (Fig. 3E). In contrast, C3 showed a significant upregulation of MYC targets v2, MYC targets v1, and E2F targets (Fig. 3F).
Fig. 3
Molecular subtyping based on the DEHRGs and DELMRGs. A Kaplan–Meier curve of the different molecular subtypes. B Heatmap of the 35 DEHRGs and DELMRGs in different subtypes and their association with clinical characteristics. C Distribution of the CMS in the molecular subtypes based on the DEHRGs and DELMRGs. D The hypoxia pathway was significantly upregulated and the oxidative phosphorylation pathway was downregulated in the C1 subtype. E The top 3 HALLMARK signaling pathways upregulated in the C1 subtype than in the C3 subtype, except hypoxia. F The top 3 HALLMARK signaling pathways upregulated in the C3 subtype than in the C1 subtype, except hypoxia. DEHRGs: different expressed hypoxia-related genes; DELMRGs: different expressed lactate metabolism-related genes; CMS: consensus molecular subtype; MAC: mucinous adenocarcinoma; NMAC: non-mucinous adenocarcinoma
Table 1 Clinicopathological characteristics of the different molecular subtypesImmune landscape of the different molecular subtypesAs hypoxia is closely related to the immune microenvironment, we evaluated immune cell infiltration in the three subtypes. We analyzed the infiltration of 22 immune cells using CIBERSORT and found that the C1 subtype predominantly contained macrophages, including M1 (p = 0.02) and M2 (p < 0.01). Meanwhile, an increase in the number of CD8 + T cells was observed in subtypes C2 and C3 (p = 0.01). Resting CD4 + memory T cells (p < 0.01), activated CD4 + memory T cells (p < 0.01), and activated dendritic cells (p < 0.01) were significantly more abundant in subtypes C2 and C3 than in the C1 subtype (Fig. 4A).
Fig. 4
Immunoscape of the different molecular subtypes. A CIBERSORT revealed the infiltration of 22 types of immune cells in the different molecular subtypes. B Differences between the three subtypes in the cancer immune cycle. C Radar chart of the differences in the recruitment of immune cells by different molecular subtypes in the cancer immune cycle. D IPS of the three molecular subtypes. E Major histocompatibility complex (MHC) molecules, effector cells (EC), suppressor cells (SC), and checkpoint (CP) scores of the different molecular subtypes. F Total TIDE scores of the three molecular subtypes. G PD-L1 and H CAF score of three molecular subtypes. I Expression of 24 well-known immune checkpoint genes in the different subtypes. IPS: immunophenoscore; TIDE: Tumor Immune Dysfunction and Exclusion; CAF: cancer-associated fibroblast. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.001
The cancer immune cycle (CIC) comprises seven major steps necessary for the immune-mediated control of tumor growth, beginning with the release of antigens from cancer cells and ending with the killing of cancer cells. The three subtypes were also evaluated for the seven major steps of CIC. The C1 subtype was found to play a significant role in the release of cancer antigens, priming and activation, trafficking of immune cells to tumors, and infiltration of immune cells into tumors (Fig. 4B). Additionally, Fig. 4C suggests that immune cells such as T cells, macrophages, and NK cells were widely recruited in the C1 subtype. However, the performance of the C1 subtype was slightly lower than that of the C2 and C3 subtypes in the recognition of cancer cells by T cells and killing of cancer cells, which are key steps in antitumor immunity, despite its ability to enrich more immune cells.
To evaluate the immunocompetence status of the three subtypes, we assessed their IPS. The IPS of the C1 subtype was significantly lower than those of subtypes C2 and C3 (p < 0.01, Fig. 4D), indicating that the C1 subtype was less responsive to immunotherapy. Specifically, the C1 subtype exhibited higher scores for MHC molecules (p < 0.01) and effector cells (p < 0.01) and lower scores for suppressor cells (p < 0.01) and checkpoints (p < 0.01, Fig. 4E). These findings appear to be inconsistent with the poor prognosis associated with the C1 subtype. To further investigate this phenomenon, we analyzed the immune functions of the samples using the TIDE algorithm. The results showed that the C1 subtype had a significantly higher overall TIDE score (p < 0.01), tumor immune dysfunction score (p < 0.01), and tumor immune exclusion score (p < 0.01) than the C2 and C3 subtypes (Fig. 4F), suggesting that immune escape was more common in the C1 subtype. Additionally, the PD-L1 score (p < 0.01, Fig. 4G) and cancer-associated fibroblast (CAF) score (p < 0.01, Fig. 4H) were significantly higher in the C1 subtype than in subtypes C2 and C3. We also evaluated the expression of 24 well-known immune checkpoint genes across the different subtypes. The results showed that these molecules, including CD274 (PD-L1), LAG3, CTLA4, and CD276, were upregulated in the C1 subtype (Fig. 4I).
Mutation mapping of the different molecular subtypesTo better understand the distinctions between the various subtypes, we examined them at the gene mutation level. Subtypes C1 and C2 exhibited a high total mutation burden (TMB), whereas subtype C3 exhibited a low TMB (p < 0.01, Fig. 5A–C). The top 20 genes in terms of mutation frequency were analyzed for each subtype. APC showed increasing mutation frequency in subtypes C1, C2, and C3 (67% vs. 72% vs. 82%), whereas MUC16 (37% vs. 30% vs. 13%) and PIK3CA (32% vs. 27% vs. 20%) showed decreasing mutation frequency in subtypes C1, C2, and C3, respectively. Additionally, TP53 mutation frequency was the highest in the C3 subtype (78%), followed by C1 (63%) and C2 (49%). The C2 subtype had the highest KRAS mutation frequency (48%), followed by subtypes C1 (40%) and C3 (33%).
Fig. 5
Mutation mapping and drug sensitivity. A–C TMB, mutation frequency and types in the three molecular subtypes. Comparison of the IC50 values of various chemotherapies in the three subtypes, including D 5-fluorouracil, E oxaliplatin, and F irinotecan. G Comparison of the AUC values of fluorouracil in the three subtypes based on the CTRP2 database. IC50: half maximal inhibitory concentration; AUC: area under the curve; TMB: tumor mutation burden
Drug sensitivityIn clinical practice, stage II CRC patients with high-risk factors and patients with stage III and IV CRC require postoperative chemotherapy. Sensitivity to commonly used chemotherapeutic agents greatly affects the outcomes and prognosis of patients with CRC. Therefore, we predicted the sensitivity of patients with the three subtypes to the most commonly used chemotherapeutic agents for CRC, namely 5-FU, oxaliplatin, and irinotecan, based on the GDSC2 and CTRP2 databases. The sensitivities of the C1, C2, and C3 subtypes to 5-FU did not differ significantly in the GDSC2 database (p = 0.51, Fig. 5D). However, patients in the C1 subtype had a significantly higher half-maximal inhibitory concentration (IC50) for oxaliplatin than those in subtypes C2 (p < 0.01) and C3 (p < 0.01); no significant difference in oxaliplatin sensitivity was found between patients in subtypes C2 and C3 (p = 0.45, Fig. 5E). There was no significant difference in the IC50 for irinotecan between subtypes C1 and C2 (p = 0.39) or C3 (p = 0.22). Patients with the C3 subtype had slightly higher IC50 values than those with the C2 subtype (p < 0.01, Fig. 5F). When we switched to the CTRP2 database, we found that the AUC for 5-FU was higher in the C1 subtype than in the C2 (p < 0.01) and C3 (p < 0.01, Fig. 5G) subtypes, suggesting that the C1 subtype was the least sensitive to 5-FU. In addition, ssGSEA found higher stemness enrichment scores in C1 (Figure S2A) and stemness-associated genes such as LGR5 and CD34 were significantly overexpressed in the C1 subtype than in the other two subtypes (Figure S2B, C). The differences in characteristics of stemness may explain the differences in drug sensitivity between the subgroups.
To gain a better understanding of the sensitivity of tumor cells to chemotherapeutic drugs under hypoxic conditions, we performed in vitro experiments under hypoxic (1% O2) and normoxic (21% O2) conditions. The experimental workflow is shown in Fig. 6A. The sensitivity of the CRC cell lines HCT116 and LS174T to the conventional chemotherapeutic agents 5-FU, oxaliplatin, and irinotecan was characterized. Moreover, the cells were treated with or without the HIF-1 inhibitor BAY87-2243 under hypoxic and normoxic conditions. Trypan blue staining revealed that the survival rate of tumor cells was higher after 48 h of hypoxia compared with that under normoxic conditions at the same concentrations of 5-FU (7.5 µM), oxaliplatin (7.5 µM), or irinotecan (10 µM in HCT116 cells and 50 µM in LS174T cells) (Fig. 6B). These results were confirmed by CCK8 experiments (Fig. 6C–H). Notably, under hypoxic conditions, BAY87-2243 increased the sensitivity of HCT116 and LS174T cells to 5-FU to normoxic levels (Fig. 6C, F). However, under both normoxic and hypoxic conditions, BAY87-2243 decreased the sensitivity of HCT116 cells to oxaliplatin (Fig. 6D); this was also observed in LS174T cells under normoxic conditions (Fig. 6G). Under hypoxic conditions, BAY87-2243 increased the sensitivity of HCT116 and LS174T cells to irinotecan (Fig. 6E, H).
Fig. 6
In vitro cell viability assay under normoxic and hypoxic circumstances. A The experimental workflow. B Trypan blue staining of cells subjected to different oxygen concentrations and treated with different chemotherapy drugs. CCK8 experiments after normoxia or hypoxia and C 5-FU, D oxaliplatin, or E irinotecan treatment combined with or without BAY87-2243 in HCT116 cells. CCK8 experiments after normoxia or hypoxia and F 5-FU, G oxaliplatin, or H irinotecan treatment combined with or without BAY87-2243 in LS174T cells. OXA: oxaliplatin; IRI: irinotecan; CCK8: Cell Counting Kit-8; ns: no significance. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.001
Construction of a prognostic prediction modelTo clarify the molecular mechanisms underlying hypoxic tumors and their impact on the OS of patients with CRC, we compared the C1 subtype, which had the worst prognosis, with the C3 subtype, which had the best prognosis. Differential expression analysis using DESeq2 revealed that 1953 genes were upregulated in the C1 subtype, while 362 genes were downregulated (Fig. 7A). The heatmap in Fig. 7B displays the top 20 genes that were upregulated and downregulated. KEGG analysis of these 2315 DEGs revealed that they were predominantly enriched in cell adhesion molecules, ECM-receptor interactions, and the PI3K-Akt signaling pathway (Fig. 7C). GO functional enrichment analysis revealed that these DEGs were significantly enriched in extracellular structure organization, extracellular matrix organization, collagen-containing extracellular matrix, and extracellular matrix structural components, suggesting a close association between DEGs and extracellular matrix (Fig. 7D).
Fig. 7
Screening process for the characterized genes. A Volcano plot of the DEGs between subtypes C1 and C3. B Heatmap of the top 20 upregulated and downregulated genes. C Top 10 KEGG pathways of the DEGs. D Top 5 pathways of the DEGs involved in GO BP, CC, and MF terms, respectively. E Venn diagram of the DEGs between subtypes C1 and C3 and genes associated with OS. F The top 20 characterized genes screened using the random forest model. G, H The LASSO regression analysis and partial likelihood deviance on the prognostic genes selected by the random forest model. DEGs: differentially expressed genes; BP: biological process; CC: cellular component; MF: molecular function; OS: overall survival; LASSO: least absolute shrinkage and selection operator
Univariate Cox regression analysis was performed to identify 2910 OS-related genes. After determining their intersection with the obtained 2315 DEGs, 830 OS-related DEGs were identified (Fig. 7E). Subsequently, the 830 DEGs were screened using a random forest model, and the top 20 genes were selected (Fig. 7F). To avoid problems with covariance and overfitting, we further screened these 20 DEGs using the LASSO method, which identified eight potential genes (Fig. 7G, H). Finally, we conducted a multivariate Cox regression analysis based on these eight genes and obtained six DEGs to construct a prognostic prediction signature, which we named the HLM score (Fig. 8A).
Fig. 8
Construction and validation of the HLM score. A Multivariate Cox regression analysis to construct the prognostic predictive model. B Distribution of risk scores, clinical events, and the expression of model genes in patients under subtypes C1 and C3. C The Kaplan–Meier survival curve for the low-risk and high-risk groups of patients in subtypes C1 and C3. D Time-dependent ROC curves for patients in subtypes C1 and C3. E Risk score distribution in subtypes C1, C2, and C3. The Kaplan–Meier survival curve for the low-risk and high-risk groups in F the total TCGA cohort and the G GSE106584, H GSE17536, and I GSE39582 datasets. J Proportion of patients who respond to anti-PD-1/L1 immunotherapy with high or low risk scores in the IMvigor210 cohort. K Kaplan–Meier survival curve for the low-risk and high-risk groups in the IMvigor210 cohort. CR: complete response; PR: partial response; SD: stable disease; PD: progressive disease; NE: not evaluated. ****p < 0.0001
Validation of the predictive efficacy of the HLM scoreWe divided the 251 patients in the C1 and C3 subtypes into high-risk and low-risk groups based on their HLM scores. A marked decrease in the OS of CRC patients was observed as the HLM score increased (Fig. 8B). The Kaplan–Meier curves indicated that patients in the high-risk group had significantly worse OS (HR = 3.14, 95% CI 1.66–5.94, p < 0.01, Fig. 8C). The AUCs of the ROC for predicting 1-, 3-, and 4-year OS were 0.78 (95% CI 0.90–0.651), 0.80 (95% CI 0.90–0.71), and 0.70 (95% CI 0.83–0.57), respectively, suggesting superior predictive efficacy (Fig. 8D). Additionally, compared with clinical factors alone, we found that when we combined the HLM score with clinical factors such as age and T, N, and M stage, it significantly improved the AUCs (Figure S3). When the HLM score was applied to the entire TCGA CRC cohort (n = 575), the C1 subtype had the highest HLM score, the C3 subtype had the lowest HLM score, and the C2 subtype had an intermediate HLM score, consistent with our previous molecular subtyping results (Fig. 8E). The TCGA CRC cohort was also divided into high-risk and low-risk groups based on the HLM scores. GSEA of the high-risk and low-risk groups revealed that the hypoxia and angiogenesis pathway were significantly upregulated in the high-risk group. In contrast, the oxidative phosphorylation pathway was significantly downregulated (Figure S4A). The expression of HIF-1α and lactate metabolism-related genes such as IGFBP3, TGFB3, and CAV1 were significantly higher in the high-risk group than in the low-risk group (Figure S4B, D). Patients in the high-risk group had a significantly worse OS (HR = 1.64, 95% CI 1.16–2.38, p < 0.01, Fig. 8F). In addition, the high-risk group, similar to the C1 subtype, exhibited a higher proportion of CMS4 while the low-risk group exhibited a higher proportion of CMS2 (Figure S5A). The effect of CMS alone on CRC prognostic stratification was insignificant. However, the HLM score in concert with CMS demonstrated the capacity to refine CRC prognostic stratification (Figure S5B, C).
To validate the efficacy of the HLM score more extensively, we obtained three datasets from the GEO database. These datasets were categorized into high- and low-risk groups based on the HLM scores. Results showed that in the GSE106584 (HR = 2.10, 95% CI 1.25–3.53, p < 0.01, Fig. 8G), GSE17536 (HR = 1.94, 95% CI 1.14–3.31, p = 0.01, Fig. 8H), and GSE39582 (HR = 1.48, 95% CI 1.09–2.00, p = 0.02, Fig. 8I) datasets, patients in the high-risk group displayed inferior OS than patients in the low-risk group. Additionally, we analyzed the IMvigor210 immunotherapy cohort, in which patients in the high-risk group had a higher proportion of progressive disease (54.42% vs. 43.48%) and fewer patients with complete response/partial response/stable disease (30.61% vs. 40.79%, p = 0.02; Fig. 8J) than the low-risk group. The Kaplan–Meier curve also suggested worse OS for patients in the high-risk group in the IMvigor210 cohort than in the low-risk group (HR = 1.36, 95% CI 1.05–1.76, p = 0.02, Fig. 8K). Besides, we assessed the efficacy of the HLM score in these four validation sets through ROC curves, risk heatmaps, and calibration curves, all of which suggest that the HLM score is robust (Figure S6).
ssGSEA and scRNA-seq analysis to assess immune cell infiltrationThe ssGSEA results indicated that the enrichment scores for CD8 + Tex, GZMK + Tex, terminal Tex, OXPHOS- Tex, and TCF7 + Tex were significantly higher in the C1 subtype than in subtypes C2 and C3 (p < 0.01, Fig. 9A). Similar results were obtained when TCGA CRC samples were categorized into high- and low-risk groups based on the HLM score (p < 0.01, Fig. 9B). To further validate these findings, publicly available scRNA-seq data were analyzed. After selection and filtering, 47,285 cells were included in the analysis. These cells clustered into five major types: B cells, T cells, myeloid cells, stromal cells, and epithelial cells (Fig. 9C, D). Based on the HLM score of each sample after pseudo-bulk analysis, the 23 CRC samples from the scRNA-seq data source were categorized into the high-risk (n = 11) and low-risk (n = 12) groups. The proportion of myeloid cells was slightly higher in the low-risk group than in the high-risk group (p = 0.05, Fig. 9E, F). We then performed subcluster annotation on 16,065T cells from these CRC samples (Fig. 9G). The proportion of exhausted CD8 + T cells (p = 0.05, Fig. 9H) and CD8 + intraepithelial lymphocytes (p = 0.04, Figure S7A) was higher in the high-risk group than in the low-risk group. To determine specific differences in the proportion of CD8 + T cells between the two groups, we performed further subpopulation annotation of CD8 + T cells (Fig. 9I). The results showed that the proportions of terminal Tex (p < 0.05, Fig. 9J), OXPHOS-Tex (p < 0.05, Fig. 9K) and GZMK + early Tem (effective memory T cells) (p < 0.05, Figure S7B) were significantly higher in the high-risk group than in the low-risk group. These findings suggest that HRGs and LMRGs may play a role in CD8 + T cell exhaustion in CRC.
Fig. 9
ssGESA and scRNA-seq analysis to assess immune cell infiltration. Enrichment scores of CD8 + Tex, GZMK + Tex, terminal Tex, OXPHOS- Tex, and TCF7 + Tex in (A) different molecular subtypes and B the high-risk and low-risk groups based on the HLM score in the TCGA cohort. C t-SNE plot of 47,285 cells from 23 patients with CRC in the GSE132465 cohort. D Percentage of the five major cell types in each sample. E Percentage of the five major cell types in the high-risk and low-risk groups based on the HLM score. F Percentage of myeloid cells in the high-risk and low-risk groups. G t-SNE plot of T cells in the GSE132465 cohort. H Percentage of CD8 + Tex in the high-risk and low-risk groups. I t-SNE plot of CD8 + T cells in the GSE132465 cohort. Percentage of J terminal Tex and K OXPHOS- Tex in the high-risk and low-risk groups. Tex: exhausted T cells; t-SNE: t-distributed stochastic neighbor embedding
In addition, we classified the 23 samples of scRNA-seq into C1/C2/C3 subtypes. It was found that, similar to the HLM score classification results, for the T cell subpopulation, exhausted CD8 + T cells especially OXPHOS- Tex were significantly higher in C1 than in C2 and C3 subtypes (Figure S7C, D). Furthermore, we found that naïve T (Tn) cells were lower in C1 than in C2, C3 subtypes (Figure S7D). Tn cells are precursors of effector and memory T cell subsets. Differences in Tn proportion between the three subtypes may be attributed to the fact that the proportion of exhausted T cells varies between the subtypes.
Establishment of a predictive nomogramA predictive nomogram based on seven factors, namely age, sex, T stage, N stage, M stage, pathological type, and HLM score, was constructed to visualize the prognosis of patients in the total TCGA CRC cohort. The nomogram could predict the OS of patients with CRC with a C-index of 0.78 (Fig. 10A). Calibration curves for the nomogram also showed ideal prediction accuracy (Fig. 10B). The DCA curve demonstrated that the nomogram provided more net clinical benefits than the clinical characteristics alone (Fig. 10C). The nomogram based on the HLM score could predict both short- and long-term OS in patients with CRC and assist their clinical management.
Fig. 10
Establishment of the prognostic predictive nomogram and single gene analysis. A Nomogram for predicting 1-, 3-, and 4-year OS using the HLM score and other clinical features. B Calibration plot showing the differences between nomogram-predicted OS and observed OS. C DCA demonstrating the net clinical benefits associated with the nomogram. D Kaplan–Meier survival curve for the high-expression and low-expression groups depending on TRIB2 expression. E TRIB2 expression in normal tissues, different molecular subtypes, and the TCGA CRC cohort. F Proteins that interact with TRIB2. G Kaplan–Meier survival curve for the high-expression and low-expression groups depending on ELFN2 expression. H ELFN2 expression in normal tissues, different molecular subtypes, and the TCGA CRC cohort. I Proteins that interact with ELFN2. DCA: decision curve analysis; MAC: mucinous adenocarcinoma; NMAC: non-mucinous adenocarcinoma; OS: overall survival; CRC: colorectal cancer
Single gene analysisTo understand how the HLM score predicts the prognosis of patients with CRC, we performed a single gene analysis of six characterized genes in C1 and C3 subtypes, and the Kaplan–Meier curve indicated that the overexpression of TRIB2 or ELFN2 predicted poorer OS in patients with CRC, with HRs of 2.39 (95% CI 1.30–4.40, p < 0.01, Fig. 10D) and 1.88 (95% CI 1.05–3.38, p = 0.03, Fig. 10G), respectively. The expression matrix revealed that TRIB2 expression was significantly higher in CRC tissues than in normal tissues. In CRC, the expression of TRIB2 increased sequentially in subtypes C3, C2, and C1 (p < 0.01, Fig. 10E). Additionally, ELFN2 was highly expressed in CRC, particularly in the C1 subtype (p < 0.01, Fig. 10H). However, TRIB2 and ELFN2 are not included in the existing set of HRGs and LMRGs. To obtain information on the proteins interacting with TRIB2 and ELFN2, we consulted the BioPlex Interactome database. Our findings indicated that ISCA1, which interacts with TRIB2, is closely related to lactate metabolism (Fig. 10F). Similarly, KLHL24, which interacts with ELFN2, is closely associated with hypoxia (Fig. 10I). Therefore, we infer that TRIB2 and ELFN2 are associated with hypoxia and lactate metabolism.
留言 (0)