
記住我

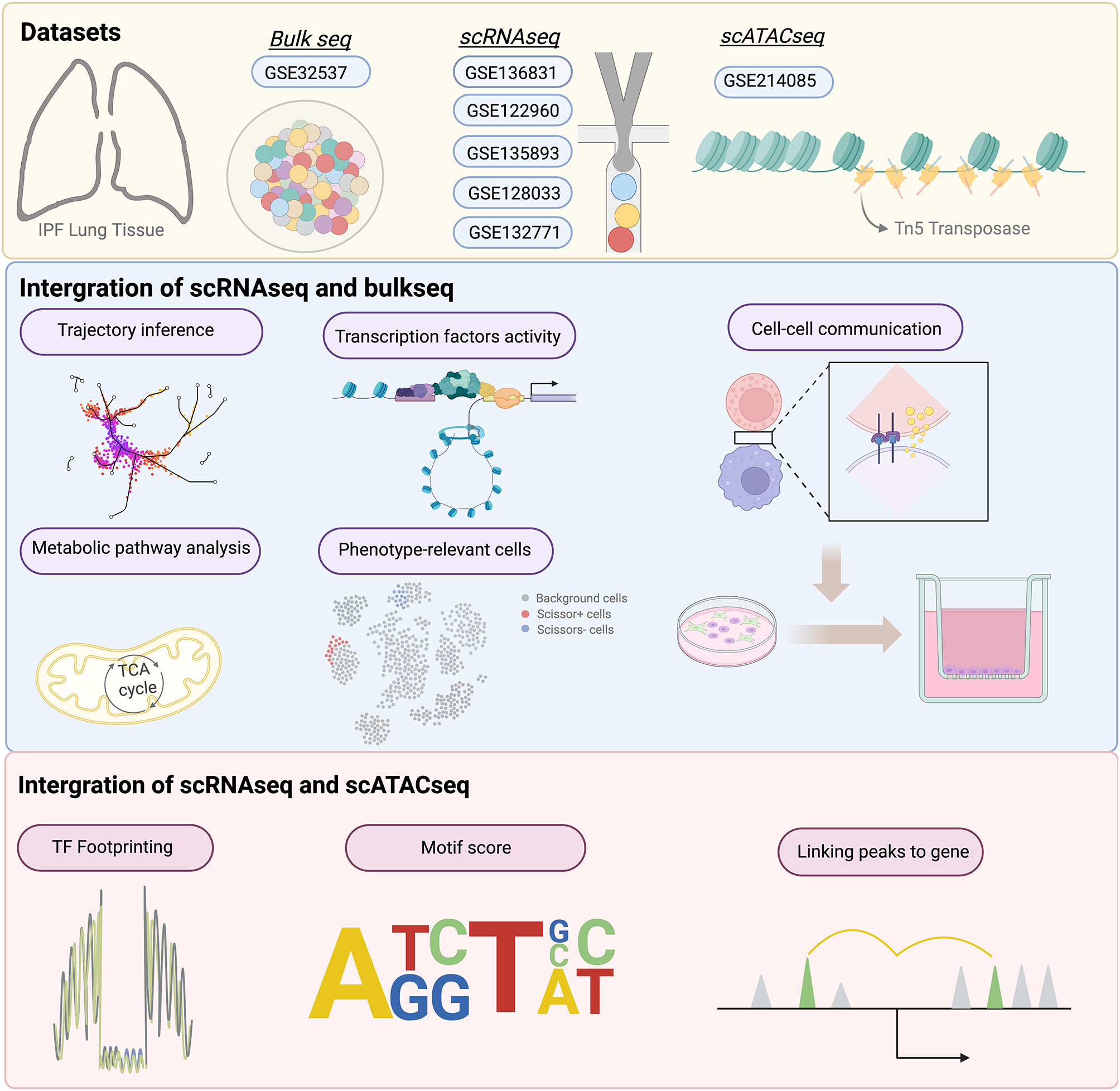


In our previous study, we developed an mNGS protocol (comprehensive mNGS, c-mNGS), allowing the detection of both DNA and RNA pathogens (including DNA viruses, RNA viruses, G + bacteria, and G- bacteria) in the samples of infectious meningitis/encephalitis (IM) in a single assay, which reduces the cost and turnaround time compared to the conventional mNGS protocols that target DNA and RNA separately [11]. In this study, we tested the performance of this protocol using a large number of CSF samples from IM patients (Fig. 1A). Briefly, 142 samples were included in this study, which were divided into three groups: CIM (n = 36), SIM (n = 43), or CTRL (n = 63) (Fig. 1A) based on traditional microbiological tests.
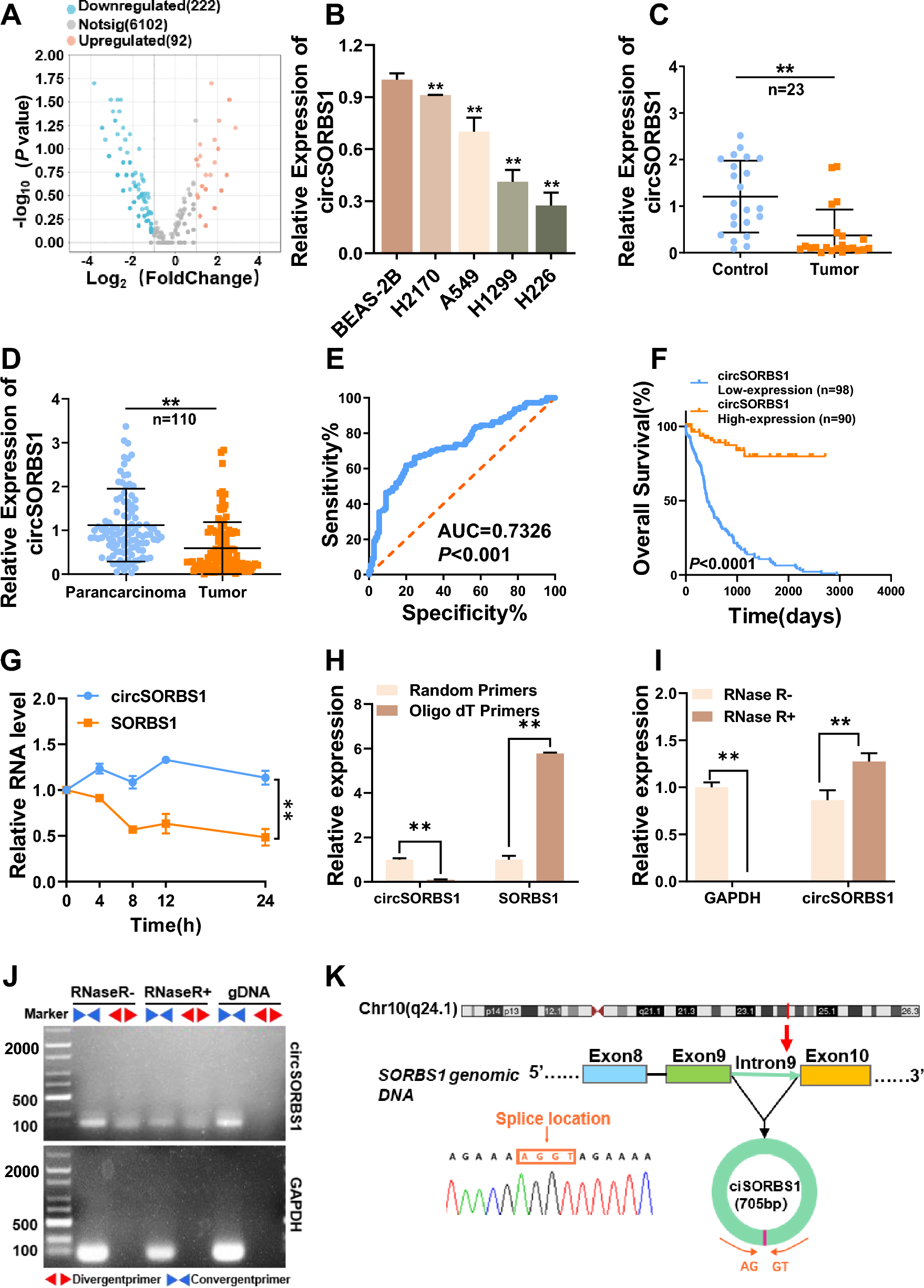
Fig. 1
The workflow of this study. (A) The groups of samples involved in this study. (B) The workflow of the study
For these samples, we generated an average of 9.30 million paired-end reads per sample (Supplementary Table 1). The kraken2 pipeline was used to align reads and identify microbial taxa as in our previous study [11, 19]. The average microbial reads from each sample was 69,082 (Supplementary Table 1). The abundance of each microbe in a sample was estimated using RPKMratio values (See methods). We optimized both methodology and pathogen reference databases to improve the accuracy of taxa identification. Using the RPKMratio as the abundance estimate and the updated reference database, our protocol can achieve AUC = 0.98 and 0.99 in the training and testing data, respectively (Fig. 2A and B). Our protocol also detected 50% more enteroviruses than the conventional protocols (Fig. 2C and D). Using RPKMratio>9.134 as the cutoff for positive prediction, our protocol can reach sensitivity and specificity of 90% and 96.6% in the training samples and 100% and 92.9% in the testing samples.
Fig. 2
Establishment of mNGS pipeline for pathogen detections. (A) The comparisons of several different normalization methods for pathogen detections in training cohorts. M1: total reads are used as normalization, M2: Total micro reads are used as normalization, M3 (RPKMratio): total micro reads and genome size are used as normalization, M4: RPKMratio: total micro reads and genome size are used as normalization without PCR duplicate removal. (B) The preformation of RPKMratio-based normalization in the validation cohort. (C) Enterovirus detected by raw database and updated database. (D) The genome coverage of Echovirus E30, which is detected by updated database only. (E) The pathogens detected by routine tests and mNGS in the same infectious meningitis and encephalitis cohorts, respectively
Additionally, our protocol can detect infections in the SIM samples where pathogens were not detected with culture or PCR in routine diagnostics (Fig. 2E). For example, HHV-7 virus was detected with 42 reads in one sample. Further, several bacterial pathogens were detected in some SIM samples with negative culture results, mainly Streptococcus pneumoniae and Ureaplasma parvum. All the newly detected pathogens using our protocol are listed in Supplementary Table 2.
Antimicrobial resistance genes of bacterial pathogens detected by mNGSThe mNGS also provides a portfolio of potential antibiotic resistance genes (ARGs) for bacterial pathogens, enhancing bacterial diagnostics and guiding treatments, and improving antibiotic stewardship [32,33,34,35].
To evaluate ARG prediction by our mNGS protocol, we started with 24 IM-positive CSF samples with > 1000 reads for bacterial pathogens. Overall, the identified ARGs are mainly associated with beta-lactam, aminoglycoside, multidrug, tetracycline, and polymyxin (Supplementary Fig. 1) and are highly heterogeneous over samples. For example, multidrug-resistant genes account for most reads in most samples. In contrast, aminoglycoside-resistant genes and beta-lactam-resistant genes accounted for more than 50% of reads in some other samples (Supplementary Fig. 1).
To check whether the identified ARGs in a sample can predict the bacteria’s antibiotic resistance, we used antimicrobial susceptibility testing as the gold standard and considered the samples with detection of Acinetobacter baumannii (AB, 5 samples), Escherichia coli (E. coli, 8 samples), and Streptococcus agalactiae (GBS, 5 samples) as these pathogens are most frequent.
In the AB samples, most ARGs are associated with antibiotics like extended-spectrum β-lactamase (ESBLs), aminoglycoside, and multidrug antibiotics (Fig. 3A). For example, blaOXA−23 and blaOXA−225 genes are found in most samples, and their presences predict resistance to commonly used ESBLs (such as IPM and MEM) (Fig. 3B). Similarly, in the E.coli samples, the presence of the ARG CTX-M co-occurs with the resistance to cephalosporin antibiotics (Fig. 3C-D). In all 3 GBS samples (12, 21, and 134) with susceptibility tests, the presence of ARGs ermB and ermC predicts resistance to Macrolide, and the presence of tetO, tetM, and tetW predicts resistance to tetracycline (Fig. 3E–F).
Fig. 3
Detections of ARGs in AB, E. coli, and GBS. (A) The ARG types and consistency of ARGs with antimicrobial susceptibility testing for AB (A-B), E. coli (C-D), and GBS (E-F), respectively. The full names of the antibiotics abbreviated in the figure are as follows: IPM (Imipenem), MEM(Meropenem), AMP (Ampicillin), CAZ(Ceftazidime), CPD (Cefpodoxime), CEZ (Ceftizoxime), CTRX (Ceftriaxone), CFPM (Cefepime), SXT (Sulfamethoxazole), TET (Tetracycline), ERY (Erythromycin) and CLI (Clindamycin)
Host response genes in infectious meningitis/encephalitisExploring the host responses can provide insights into both the diagnosis and prognosis of IM. Our c-mNGS protocol measures both DNA and RNA at the same time and thus provides the ability to profile host gene expressions (Fig. 1B).
To identify differentially expressed genes, we compared the samples of bacterial meningitis (BM; 47 samples) and the control (CTRL; 37 samples). We identified 1036 DEGs (Supplementary Table 3) and found 48 enriched KEGG pathways by the GSEA method. The top terms include oxidative stress (hsa00190: Oxidative phosphorylation and hsa05208: Chemical carcinogenesis-reactive oxygen species) and antigen processing (hsa04612: Antigen processing and presentation) and immune responses (hsa05332: Graft-versus-host disease and hsa05320:Autoimmune thyroid disease) (Fig. 4A).
Fig. 4
Functional enrichment analysis, classification model, and biomarkers for IM. (A) The enriched KEGG pathways between BM and CTRL samples. (B) The enriched KEGG pathways between VM and CTRL samples. (C) The t-SNE visualization results for BM, VM, and CTRL. (D-E) The expressions of BM-associated genes (D) and VM-associated genes (E)
Similarly, 26 viral meningitis/encephalitis (VM) samples were compared to the control. Similarly, we identified 22 VM-vs-CTRL DEGs (Supplementary Table 3) and found 40 enriched KEGG pathways by the GSEA method. The top 10 pathways are shown in Fig. 4B, including immune rejection (hsa05330: Allograft rejection, hsa05332: Graft-versus-host disease and hsa04612: Antigen processing and presentation), and viral infections (hsa05168:Herpes simplex virus 1 infection and hsa05169: Epstein-Barr virus infection).
Finally, we tried to obtain several host genes that can distinguish BM, VM, and CTRL samples. The R package DaMiRseq was used to rank and select the most robust genes for the model (See methods), and 53 genes were obtained. The genes can separate the samples very well (Fig. 4C). And based on the 53 genes, we built a logistic regression 3-class model which showed high classification performance and achieved AUC values of 0.972, 0.967 and 0.994 for BM, VM and CTRL, respectively (Fig. 4D). The genes associated with the scores of BM and VM in the classification model can be found in Supplementary Fig. 2 and some genes are knowingly associated with infections, such as ASRGL1, NR2F6, and OLFML3 for bacterial infection (Fig. 4E) and STIP1, PGAM5, and AKAP8 for viral infections (Fig. 4F).
Using host gene expression response to detect bacterial contaminationsBacterial contaminations are widespread for CSF samples, leading to false-positive diagnoses and costly, possibly unnecessary treatments [36]. One strategy to identify potential contaminations is to examine host gene expression in a CSF sample because they are unlikely affected by contamination. To this end, we developed a BM/CTRL classification model based on host gene profiling (Fig. 5A).
Fig. 5
IM classification model can identify false-positive mNGS results caused by contamination. (A) The diagram of the host-pathogen combined method for contamination identification. (B) The performances of the BM/CTRL classification model. (C) The results of mNGS for 3 CSF samples with suspected contamination. (D) The results of the BM classification model for the above 3 CSF samples
We randomly divided 82 samples (including BM and CTRL subjects) into training (n = 54) and test cohorts (n = 28). The top five differentially expressed genes between BM and CTRL samples were selected via the DaMiRseq package (See methods) and were used to develop a logistic regression model (LRM). The model performed well in both training (AUC = 0.947, sensitivity = 90.6%, and specificity = 86.4%) and test cohorts (AUC of 0.969, sensitivity = 93.8%, and specificity = 83.3%) (Fig. 5B), providing a tool to rule out contaminations.
Candidate pathogens were identified in 3 CSF samples by mNGS reads (Fig. 5C), which may be subject to contaminations. By applying the model to these 3 CSF samples, we found that all the samples are infection-free (Fig. 5D). These results are in line with the observation that these samples are near-normal in biochemical indicators and clinical manifestations.
Developing a model to identify BM patients with poor prognosisAccording to the outcomes when discharged, more than half of BM patients (54.9%, 28/51) had poor prognosis. Poor prognosis is associated with complications of bacterial meningitis (including subdural effusion, ependymitis, hydrocephalus, encephalomalacia, and brain abscess), withdrawal of treatment, or death [37]. To predict prognosis, we developed a model based on ten differentially expressed genes between good and poor prognosis groups selected via the DaMiRseq algorithm (Fig. 6A). The BM samples were randomly divided into training (n = 33) and test cohorts (n = 18). And a logistic regression was trained from it. Finally, four genes, including CXXC4, XPNPEP2, IGSF1 and ND4L, were used in the model (Fig. 6B). As seen in Fig. 6C, the model performs well in both training ((AUC = 0.88, sensitivity = 86.7% and specificity = 88.9%) and test cohorts (AUC = 0.78, sensitivity = 75% and specificity = 80%).
Fig. 6
The logistics regression model for predicting BM prognosis. (A) The expressions of poor prognosis-related genes. (B) The performance of the BM prognostic risk prediction model in training and validation cohorts. (C) The expressions of CXXC4, XPNPEP2, IGSF1, and ND4L genes in poor and good prognosis samples
留言 (0)