2.1 Patients
From October 2010 to December 2012, 212 primary CRC tissues and 47 matching adjacent noncancerous tissues were obtained from patients who had the operation at the Hunan Provincial People’s Hospital. Patients with preoperative chemotherapy and/or radiotherapy, and palliative surgery were excluded. Survival analysis excluded patients who died within 30 days after surgery, since their death could be attributed to surgical complications. All patients have signed an informed consent. The Ethical Review Board of Hunan Provincial People’s Hospital have approved the study. The study complied with the guidelines of the Declaration of Helsinki.
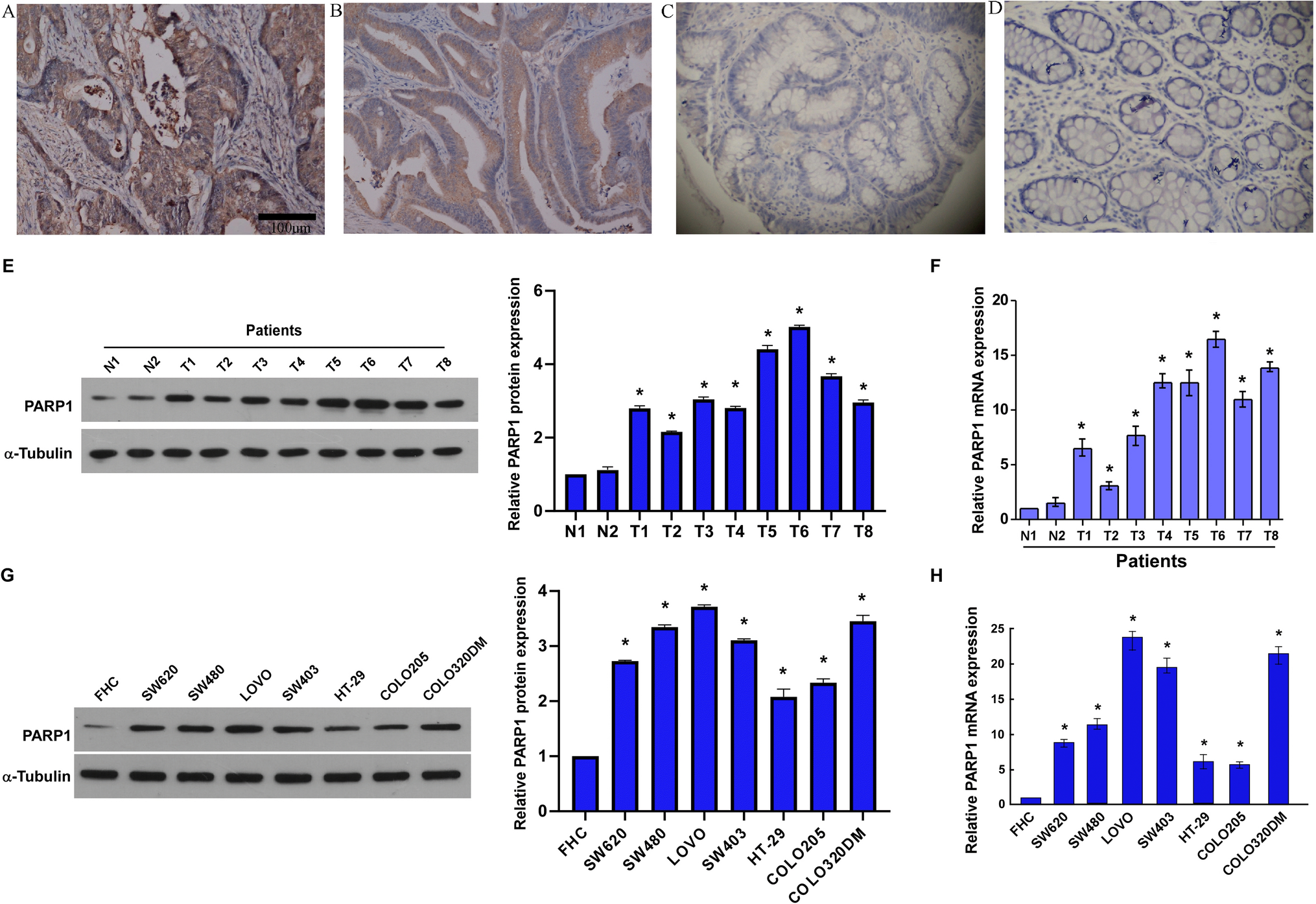
2.2 Immunohistochemistry (IHC)
According to previously described methods, we performed the following procedures by the classic biotin–streptavidin–peroxidase IHC staining protocols [15]. We obtained sections from the Hunan Provincial People’s Hospital Pathology Department, and incubated overnight at 48 °C with polyclonal primary antibody against PARP1 (1:100; Abcam, Cambridge, UK). Following incubated with diaminobenzidine and horseradish peroxidase-conjugated sheep anti-rabbit secondary antibody (Beyotime; Guangzhou, China), used Mayer’s hematoxylin to counterstain the slides. Positive control was primary CRC tissue section. In negative control staining, used phosphate-buffered saline (PBS) buffer instead of primary antibody. The immunostaining results were scored according to the methods previously described below [16].
2.3 Culture and treatment of cell
We obtained human CRC cell lines FHC, SW480, SW620, LoVo, SW403, HT-29, COLO205 and COLO320DM from the American Type Culture Collection (Manassas, VA, USA). Incubated cell lines in DMEM/RPMI-1640 medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and 10% FBS (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C with 5% CO2.
2.4 Vectors, retroviral infection and transfection
Through subcloning the PCR-amplified human PARP1 coding sequence into a pBABE-puro vector, we generated pBABE/PARP1-overexpressing human PARP1. Cloned two RNA interference (RNAi) oligonucleotides into pSuper-retro-puro vectors to produce the pSuper-retro-PARP1-RNAi respectively, thereby silencing endogenous PARP1. As mentioned earlier, the generation and infection of retroviruses were carried out to establish stable cell lines [17]. The calcium phosphate transfection method was used to cotransfect the retroviruses into 293FT cells and then harvested and infected cells. After infection 48 h, cell line stably overexpress of PARP1 and the vector control name as (SW480/PARP1; SW480/vector, SW620/PARP1, SW620/vector, respectively) or PARP1 RNAi and the Negative control (SW480/PARP1/RNAi, SW480/Scramble, SW620/PARP1/RNAi; SW620/Scramble, respectively) were selected using puromycin (0.5 mg/mL) over 10 days. SDS-PAGE was used to segregate SW480 and SW620 cell lysates to detect PARP1 protein levels, and the cell proliferation was analyzed with the stable cell line. The XRCC2 inhibitor (XRCC2/siRNA), negative control (NC) were purchased from RiboBio Co. Ltd (Guangzhou, Guangdong, China). Transfection of oligonucleotides was performed using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA), according to the manufacturers’ protocol.
2.5 Extraction and reverse transcription of RNA, real-time quantitative PCR
In line with manufacturer’s illustrations, applied Trizol reagent (Invitrogen, Carlsbad, CA, USA) to accomplish total RNA extraction from cultured cells or tissues. cDNAs were amplified and equipped with the ABI PRISM 7500 system (Applied Biosystems, Foster City, CA, USA), SYBR Green I (Invitrogen, Carlsbad, CA, USA) was applied for real-time quantitative PCR. Selected housekeeping gene GAPDH as an internal control. Used the primers below: PARP1 forward, 5′-ACAGTGTGCAGGCCAAGGTG-3′, and reverse 5′-CTCGGC TTCTTCAGAATCTCTGTC-3′; XRCC2 forward: 5′-TCACCTGTGCATGGTG ATATT-3′, and reverse: 5′-TTCCAGGCCACCTTCTGATT-3′; GAPDH forward: 5′-GACTCATGACCACAGTCCATGC-3′, and reverse: 5′-AGAGGCAGGGATGATG TTCTG-3′; p21 forward: 5′-CGATGCCAACCTCCTCAACGA-3′, and reverse: 5′-TCGCAGACCTCCAGCATCCA-3′; cyclin D1 forward: 5′-AACTACCTGGA CCGCTTCCT-3′, and reverse: 5′-CCACTTGAGCTTGTTCACCA-3′. Genes expression data were normalized to the geometric mean of housekeeping gene GAPDH to control the variability in expression levels and calculated as 2−[(Ct of gene) − (Ct of GAPDH)], where Ct represents the threshold cycle for each transcript.
2.6 Western blotting
According to manufacturer’s instructions, proteins were prepared from cell lysates, BCA method for determining protein content; isolated on SDS-PAGE, and transferred to PVDF membranes. PVDF membranes were cut prior to hybridisation with antibodies during blotting. To detect specific proteins, primary antibodies that were used included α-Tubulin mouse monoclonal antibody (1:1000; Sigma-Aldrich, St. Louis, MO, USA), anti-human XRCC2 mouse monoclonal antibody (1:1500; Abcam), anti-human PARP1 mouse monoclonal antibody (1:1500; Abcam), anti-human cyclin D1 rabbit monoclonal antibody (1:1500; Abcam), and anti-human P21 rabbit monoclonal antibody (1:1500; Abcam). The secondary antibody was goat anti-mouse antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After the membrane incubated with Clarity enhanced chemiluminescence (ECL), and we cut the X-ray film to a suitable size for development. An anti-α-tubulin antibody was used as a loading control. At least three biological replicates were used to detect the sample.
2.7 Cell proliferation detection
In accordance with the manufacturer’s illustrations, used the Cell Counting Kit-8 (CCK-8) cell proliferation kit (Dojindo Laboratories, Kumamoto, Japan) to assess cell proliferation. Concisely, seeded the cells into 96-well plates (2 × 103 cells/well), and cultured under regular circumstances with 100 μL complete medium. At the specified time, incubated cells with RPMI-1640 medium (100 μL) plus CCK8 reagent (10 μL) for 2 h at 37 °C. After that, measured the absorbance at 450 nm wavelength on a microplate reader (Bio-Rad, La Jolla, CA, USA). Conducted three repetition experiments independently.
2.8 Colony formation assay
In brief, plated exponential growth cells into 6-well plates at 1000 cells/well and cultured for 10–14 days at 37 °C with 5% CO2. For visualization and counting, used 75% ethanol to fix the colonies for 30 min and stained with 0.5% crystal violet (Beyotime, Nanjing, China) afterwards. When colonies with more than 50 cells would be manually calculated. Every group of cells comprised three wells, and three independent repeat experiments were conducted.
2.9 Co-immunoprecipitation (Co-IP)
Co-IP was carried out using Pierce Co-Immunoprecipitation Kit (cat. 26149, Thermo Fisher Scientific, Waltham, MA, USA) according to the protocol provided by the manufacturer. SW620 cells and anti-human XRCC2 mouse monoclonal antibody (1:1500; Abcam, Cambridge, MA, USA), anti-human PARP1 mouse monoclonal antibody (1:1500; Abcam, Cambridge, MA, USA) were used.
2.10 Statistical analysis
Statistical analysis was conducted using SPSS 20.0 (SPSS Inc, Chicago, IL, USA). Employed the Chi-square test to evaluate the association between PARP1 expression and clinicopathological characteristics. The significant differences between two groups of data were analyzed with the Student’s t test. The log-rank test and the Kaplan–Meier method were employed for survival curves analysis. The time from surgery to last follow-up date or patient’s death was 5-year overall survival (OS). The time from curative surgery to recurrence, the final follow-up date, or death was defined as Relapse-free survival (RFS). Local and distant relapses were regarded as recurrence. The statistical significance was set at p < 0.05.






留言 (0)