
記住我



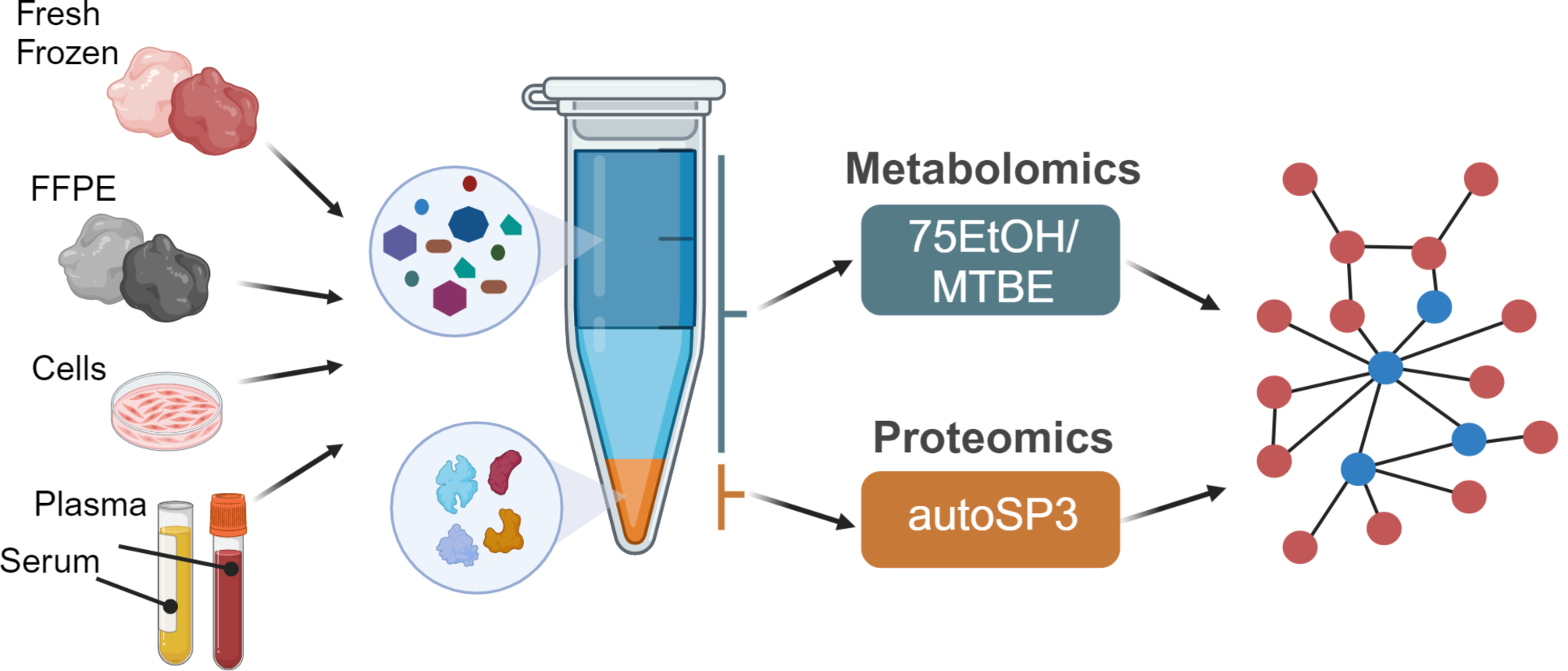
Early disease detection relies on the identification and quantification of reliable biomarkers. The pooled human plasma samples were divided into 8 aliquots, and each aliquot underwent processing to evaluate the Neat Plasma workflow and commercially available sample preparation kit ENRICH-iST and the fully automated Proteograph XT workflow, as depicted in (Fig. 1). The Neat Plasma workflow entails manual processing with laboratory reagents, while the ENRICH-iST approach enriches proteins, providing a streamlined sample preparation workflow. In contrast, the Proteograph XT workflow is fully automated and utilizes two nanoparticles, selectively enriching an unbiased subset of proteins in complex plasma samples.
Fig. 1
Plasma sample preparation workflows. A visual representation comparing the Neat Plasma, ENRICH-iST, and Proteograph XT workflows step by step.
All three workflows (Fig. 1) were processed using identical pooled plasma aliquots, and data acquisition was conducted on a timsTOF Pro 2 instrument with a 65 min gradient and diaPASEF method. The subsequent data analysis was performed employing DIA-NN. Initially, the protein identification performance of each workflow was assessed. Across all three workflows, approximately 5881 protein groups were identified. Notably, Proteograph XT exhibited superior performance, identifying, and quantifying over 4.2-fold more protein groups compared to Neat Plasma and 2.4-fold more compared to ENRICH-iST (Supplementary Information: Table S1, Fig. 2A). Similarly, 66,987 peptides were identified, with Proteograph XT quantifying over 6.7-fold more compared to Neat Plasma and fourfold more compared to ENRICH-iST (Supplementary Information: Table S2, Fig. 2B).
Fig. 2
Plasma sample preparation workflow comparison. A Protein groups identified by each workflow B Peptides identified by each workflow. C Coefficient of Variation (CV) for median quantified protein intensities within workflows. D The dynamic range of quantified protein abundance across workflows
The protein dynamic range and complexity play crucial roles in the depth of the quantified plasma proteome, with Neat Plasma samples providing the least information. However, ENRICH-iST exhibits improvement compared to Neat Plasma, and the Proteograph XT workflow outperforms both alternatives.
Large cohort studies rely on a robust and reproducible workflow. We compared the quantified normalized intensity of protein groups within different workflows Neat Plasma, ENRICH-iST, and Proteograph XT. The Neat Plasma, ENRICH-iST, and Proteograph XT workflows yielded a median coefficient of variation (CV) of 24.6, 21.0, and 10.7%, respectively, as shown in (Fig. 2C). The Proteograph XT workflow demonstrated the lowest CV compared to the Neat Plasma and ENRICH-iST workflows, attributed to the uniform and consistent enrichment of proteins using SeerProteograph's nanoparticle technology, operating across a large dynamic range. The fully automated capabilities of SeerProteograph also contribute to minimizing technical challenges in the workflow.
Plasma/serum samples are complex due to the broad dynamic range of proteins, posing challenges for the identification and quantification of low-abundant proteins through LC–MS/MS. To assess the dynamic range covered by each workflow, we utilized a protein abundance ranking of protein groups' normalized intensities, revealing an approximate span of 4.6 orders of magnitude. The Proteograph XT workflow significantly increased the number of quantified proteins by over 6.3-fold and 3.4-fold compared to the Neat Plasma and ENRICH-iST workflows. This extension indicates a highly efficient reduction of the dynamic range (Fig. 2D) compared to the Neat Plasma and ENRICH-iST workflows.
Comparative analysis of workflows for secretome database coverageNext, we explored the coverage of the secretome database, which comprises soluble proteins and secreted extracellular vesicles, encompassing biologically active factors such as cytokines, interleukins, interferons, chemokines, complement and coagulation factors, hormones, growth factors, enzymes [22]. These proteins, shed from cells/tumors, play a crucial role in cell signaling, communication, and growth, and their abundance changes under various pathological conditions. While these proteins are secreted into the extracellular space, they are generally more abundant in biological fluids [23]. The dynamic nature of secretome protein composition makes them a valuable source of potential biomarkers for cancer and other diseases, aiding in diagnosis, prognosis, and therapeutic monitoring [24].
The Secretome database, sourced from The Human Protein Atlas [25], underwent a comprehensive comparison across the Neat Plasma, ENRICH-iST, and Proteograph XT workflows to assess coverage. Proteins quantified in all samples within these workflows were included in the analysis, revealing that the Proteograph XT workflow exhibited notably high coverage, particularly in the quantification of low-abundant proteins (Fig. 3A).
Fig. 3
Secretome Protein Database Coverage. A Evaluation of protein coverage from the Secretome protein database across workflows. B Assessment of the percentage overlap between SeerProteograph and Secretome database protein groups. C Gene Ontology (GO) enrichment, and D KEEG pathway analysis for protein groups overlapping between Proteograph XT and Secretome database.
For Gene Ontology (GO) terms functional analysis, a ~ 39% overlap of proteins of Proteograph XT workflow was chosen (Fig. 3B). This analysis encompassed Molecular Function (MF), Biological Processes (BP), and Cellular Compartments (CC) (Fig. 3C). The proteins predicted to be secreted into human blood encompassed a diverse array, including well-characterized proteins associated with the extracellular matrix organization, enzymes, receptors, cytokines, complement activation, peptidase activator, humoral immune response, wound healing, leukocyte migration, cell chemotaxis, myeloid leukocyte migration, transport proteins, developmental proteins, defense proteins, enzymes, enzyme inhibitors, integrin binding, antigen binding, glycosaminoglycan binding, collagen binding, B cell-mediated immunity-related proteins, and classical pathway.
While the identified proteins were found in plasma, statistical analysis suggests they are secreted from various cellular compartments, including the endoplasmic reticulum (ER) lumen, vesicle lumen, secretory granule lumen, blood microparticles, lysosomal lumen, platelet alpha granule lumen, Golgi lumen, plasma lipoprotein particles, and protein-lipid complexes.
In the KEGG pathway analysis, these proteins showed significant enrichment for a variety of pathways including complement and coagulation cascades, cytokine-cytokine receptor interaction, PI3K-AKT signaling pathways, ECM-receptor interaction, lysosome, protein digestion and absorption, cholesterol metabolism, TGF-beta signaling pathway, antigen processing and presentation, fat digestion and absorption, glycosaminoglycan degradation pathways (Fig. 3D).
Comparative analysis of workflows for functional annotation coverageWe investigated the coverage of proteins quantified in three workflows using functional annotation enrichment analysis. Hierarchical clustering of quantified proteins based on their log2 intensity yielded three distinct groups of clusters (Fig. 4A). Each cluster was analyzed for enriched pathways using ClusterProfiler R package of the function of compareCluster with WikiPathways [20] using a threshold of Benjamini and Hochberg (BH) adjusted p-value < 0.05. Proteins covered with cluster 1 showed significant enrichment for a variety of pathways including complement and coagulation cascades, complement system, complement activation, blood clotting cascade, lipid particle composition, cholesterol metabolism, metabolism of triglycerides, and acute inflammatory response. Proteins present in Cluster 1, quantified in all three workflows, these proteins are highly abundant and consistently quantified.
Fig. 4
Functional Annotation Coverage. A Hierarchical clustering of normalized Log2 protein intensities B Pathway analysis of enriched proteins in each identified cluster using the ClusterProfiler package
Proteins associated with EGF EGFR signaling, VEGFA VEGFR2 signaling, glycolysis and gluconeogenesis, chemokine signaling pathway, and B cell receptor signaling pathway are enriched by cluster 2. Proteins present in Cluster 2, quantified in ENRICH-iST, and Proteograph XT workflows.
Proteins associated with Insulin signaling, TNF alpha signaling pathway, T and B cell receptor signaling, IL1/2/5 signaling, proteasome degradation pathways were enriched by cluster 3. Cluster 3 proteins were identified in Proteograph XT workflow only, these proteins are low abundant in the samples and could potentially serve as crucial biomarkers.
留言 (0)