
記住我
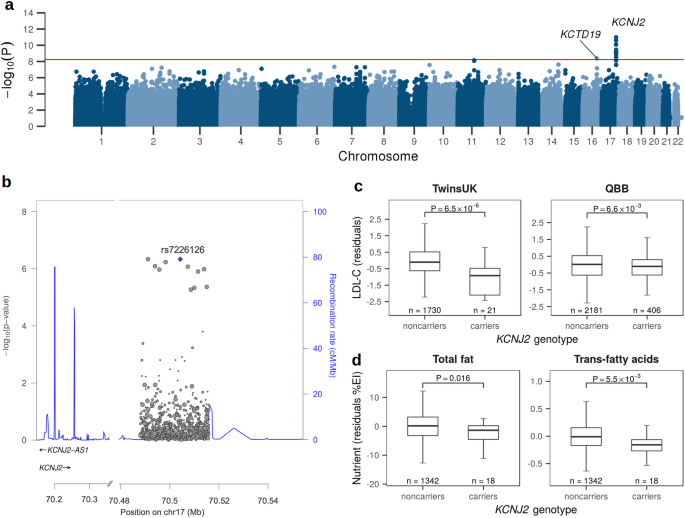



To assess the presence of HPV DNA in Vulvar Squamous Cell Carcinomas (VSCCs), genomic DNA was extracted from 13 vulvar tumors, all of which were VSCCs (Table 1). The initial screening involved PCR amplification using primers (GP5+/GP6+) that target a conserved region within the viral L1 gene, shared by multiple mucosotropic HPV types22. The overall median age of the cohort was 67.07 years [range: 43–85] and included various self-reported ethnicities. Six of the 13 tumors (46%) had recurrent disease at the time of biopsy, and three samples (23%) were from women that received neoadjuvant radiotherapy and chemotherapy prior to tumor biopsy. DNAs were extracted from 12 primary vulvar tumor sites, and 1 metastatic site biopsy (Tumor 2). HPV DNA was detected in tumors 2, 4, 5, and 10 (31% of the samples) (Supplementary Fig. 1), indicating the presence of HPV DNA in the tumors, but not whether the viral DNA was present in episomal or integrated form (Table 2).
Table 1 Clinical characteristics of patients and vulvar tumors included in the studyTable 2 HPV status, HPV type, and integration sites detected in VVSCComprehensive HPV type identification and characterization of integrated HPV DNA using HC + SEQSince the breadth of effectiveness of GP5+/GP6+ primers with less common HPV types is uncertain22, and some HPV-induced cancers contain integrated, subgenomic HPV DNA segments lacking the GP5+/GP6+ target segment12, we performed HC + SEQ on all 13 samples to define the identity and structure of HPV DNA in each21. HPV DNA enrichment was accomplished through the utilization of a custom-designed capture probe set, consisting of oligonucleotide probes complementary to 143 different HPV types (HPV1 through HPV14310). This comprehensive probe set encompasses the high-risk types recognized by the World Health Organization (WHO) International Agency For Research on Cancer (IARC) as carcinogenic or probable carcinogenic (HPV-16, -18, -31, -33, -35, -39, -45, -51, -52, -56, -58, -59, -68, -69)23. Biotinylated DNA probes, each comprising overlapping, single-stranded sequences covering both DNA strands of the full ~8 kbp genomes of each HPV type, allowed capture along the entire viral genome of each sample if present. The paired-end sequencing reads obtained were aligned using STAR24,25 to a combined reference genome comprising the GRCh38/hg38 human genome assembly plus the sequences of all 143 HPV types included in the capture panel based on reference genomes in the Papillomavirus Episteme10.
HC + SEQ detected HPV DNA in five samples (39%) the four that tested positive by PCR using the GP5+/GP6+ primers and one additional tumor (Tumor 13). Average HPV genome coverage by HC + SEQ ranged from 139X to 8414X (Supplementary Table 1). Alignment of sequence reads across the viral genome (Fig. 1) allowed unambiguous identification of the HPV types in each of the five tumors, illustrating an inherent strength of the HC + SEQ approach. Tumors 2, 4 and 5 yielded DNA exclusively from HPV16, the most prevalent of the high-risk HPVs in human cancers independent of tumor anatomical site. In tumor 2, HPV16 genome read coverage was sufficiently high to determine that the predominant HPV DNA comprised a segment of about three-fourths of the viral genome extending from a carboxyl terminal-encoding portion of L1, through the URR, E6, E7, E1, and into an amino terminal-encoding portion of E2, with the segment from reference genome positions 3582 to 5588 absent. In addition, coverage of full-length HPV16 DNA was also detected in this tumor, although at much lower levels, suggesting that only a small fraction of cells in the tumor contained the full-length HPV16 genome. Tumor 4 contained a near full-length sequence of HPV16 DNA, missing a 16 bp segment from reference genome positions 4162 to 4178 in the E5 gene. Tumor 5 yielded reads mapping to the complete HPV16 genome.
Fig. 1: Coverage of HPV genomes and typing based on viral DNA hybridization capture plus Illumina short read sequencing.
The horizontal scale depicts the linearized HPV genome, starting from E6 (positions 1 and 7906 in HPV16). The corresponding locations of viral open reading frames are displayed at the bottom. The plot depicts the deduplicated DNA sequence read counts for each sample, with the Y-axis scales on the left for each tumor. The identified HPV type(s) are indicated on the right side of the plot. The boundaries of underrepresented segments in Tumor 2 (3582–5588), Tumor 10 HPV16 component (URR-E6-E7 segment 7135 to 1805), and Tumor 13 HPV53 component (4825 to 5297) are indicated.
In contrast, Tumors 10 and 13 both contained two HPV types, with substantial DNA sequence read count coverage for each type, indicating that both patients had incurred co-infections (Fig. 1). Tumor 10 contained DNA covering the complete genomes of both HPV6, considered low risk for cervical cancer, and high-risk HPV16. Likewise, Tumor 13 contained two HPV types, HPV53 and HPV62, both relatively uncommon HPV types with neither currently categorized as group 1 carcinogenic or group 2 probable carcinogenic23. However, it is worth noting that HPV53 has been previously associated with cancer cases26. The HPV53 DNA comprised a subgenomic segment with loss of viral DNA sequence from positions 4825 to 5297 in the L2 gene, while the entire HPV62 genome was present. The number of read counts for HPV53 was about 16-fold higher than for HPV62 (Supplementary Table 1), suggesting that the former is likely more abundant, and that it was more likely to have had a carcinogenic role than HPV62. These data do not differentiate whether DNA from the two HPV types were within the same cells, or if each type existed in distinct, separate cells in the tumors. Nonetheless, the detection of abundant DNA of two relatively uncommon HPV types in a tumor that was positive by HC + SEQ but not by GP5+/GP6 + PCR illustrated the breadth of sensitivity of the contemporary HC + SEQ approach to detect HPV in tumors. In summary, HPV DNA was detected in five of the tumors, and in all these cases, the identified DNAs retained the upstream regulatory region (URR), as well as the open reading frames (ORFs) of E6 and E7 of the cognate HPV types.
HPV DNA is integrated into the human genome in some vulvar cancersTo determine whether HPV DNA was integrated into the human genome in the five HPV-positive vulvar tumors, the DNA sequence reads were computationally examined for split reads containing a junction between HPV and human DNAs using STAR24,25. Junctions were detected in Tumors 2, 4 and 5 (Fig. 2, junction sequences shown in Supplementary Fig. 2). In Tumor 2, HPV16 was integrated into the first intron of the Nck-associated protein 1 gene (NCKAP1) gene on chromosome 2, with the viral genome oriented in the opposite transcriptional direction as NCKAP1 (Fig. 2a). The HPV-human DNA junctions were at HPV positions 3582 and 5588, the precise base pairs at the ends of a high coverage segment in Fig. 1 and were joined to intron 1 of NCKAP1. Thus HC + SEQ showed that the predominant form of HPV16 in this tumor was an integrated, subgenomic viral DNA segment between those positions. Moreover, a 9 bp duplicated sequence (5′-CAACACGGT-3′) immediately flanked both sides of the viral insertion (Fig. 2a, Supplementary Fig. 2). In Tumor 4, HPV16 was integrated into chromosome 5 in the second intron and in the opposite transcriptional orientation of Chromosome 5 Open Reading Frame 67 (C5orf67) (Fig. 2b), a gene of unknown molecular function. The viral DNA ends at the two junctions again occurred precisely at the ends of the deletion determined by HC + SEQ (16 bp, positions 4162 to 4178 in the E5 gene, Fig. 1). The insertion was flanked by a direct repeat of 3 bp (5′-TTT-3′) from the human genome (Fig. 2b, Supplementary Fig. 2). In Tumor 5, HPV16 DNA was integrated into the LDL receptor-related protein 1B (LRP1B) gene on chromosome 2 (Fig. 2C). The viral DNA was again in the opposite transcriptional orientation as the human gene, with the junctions in introns 11 and intron 8 of LRP1B, leading to the inference that integration was accompanied by a 38 kbp deletion between those introns including exons 9 through 11 (Fig. 2c). Each of the junctions had a short sequence identity (microhomology) between the cognate ends of the human and HPV16 genomes (1 bp or 8 bp, Supplementary Fig. 2), suggesting joining of viral and human DNAs by microhomology-mediated end joining17. Such microhomologies or insertions occur frequently at HPV DNA insertion junctions in cervical tumors27. Since the entire HPV16 genome was covered by sequence reads, we were able to infer that the inserted HPV16 DNA comprised a tandem structure longer than one 7906 bp viral genome (Fig. 2c). All HPV-human DNA junctions were confirmed by PCR on tumor DNAs (Supplementary Fig. 2).
Fig. 2: Structures of integrated HPV16 DNAs in Tumors 2, 4, and 5 based on HC + SEQ aligned at the URR-E6-E7 segment.
For each tumor, the approximate position of the HPV DNA insertion (red bar) is shown on the cognate chromosome ideogram. The structure of each integrated DNA is represented as a thick blue arrow, with viral DNA segment length and the approximate positions of viral open reading frames (ORFs) and the upstream regulatory region (URR) also shown in blue. Flanking the viral DNA junctions, human genome segments are depicted as thinner green arrows, including the adjacent flanking exons shown as rectangles. Arrows indicate the 5’ to 3’ orientation of the plus strands of both viral and cellular genes, with the HPV16 DNAs integrated into the opposite transcriptional orientation compared to the human genes in each of the three tumors. The positions of the insertion junctions in the human and HPV reference genome (hg38 and PAVE, respectively) are indicated in green or blue, respectively. For Tumors 2 and 4, short, direct repeat, human DNA sequences immediately flanking the insertions are shown in red. In Tumor 5, segments of microhomology between the viral and human DNA at the junctions are depicted as a short red line. Below the genomes for Tumors 2 and 5, positions of long-range DNA sequence reads are represented by dotted black arrows. HPV and human DNAs were drawn at different scales. a Genetic diagram illustrating the structure of the HPV16 DNA segment integrated in intron 1 of the NCKAP1 gene in Tumor 2, with the 9 bp, direct repeat sequence (5′-CAACACGGT-3′) immediately flanking both sides of the viral insertion in red. b Genetic diagram of the HPV16 segment in intron 2 of the C5orf67 gene, where a 3 bp direct repeat sequence (5′-TTT-3′) is shown immediately flanking the viral insertion on both sides in red. c Genetic diagram of HPV16 DNA in Tumor 5 inserted between exons 7 and 12 in the LRBP1 gene. The HPV16 genome was noted to be present in tandem with at least one full genome and an additional 982 bp, as determined by HC + SEQ and long-range MinION nanopore sequencing reads spanning this region. This integration event coincided with a 38 kbp deletion within the LRBP1 gene. d Long-range MinION nanopore sequencing of Tumor 4, specifically showing the sequence read coverage for a 62 kbp segment from the second intron of C5orf67 on chromosome 5. The plot depicts the over-representation of a 28 kb segment containing the HPV16 DNA segment. Below the read count plot, a linear genetic diagram represents the inserted HPV16 DNA and the 28 kb stretch of high read counts. Circular episome and DNA concatemer structures are depicted below, each consistent with the sequencing results.
To analyze the structures and junctions further, long-range DNA sequencing was also performed on genomic DNA from each of the three tumors with integrated HPV16 DNA. While prone to short sequence misreads and indels, this technique is effective for elucidating larger scale genomic structures and rearrangements. One long sequence read was obtained confirming the insertion in one allele of NCKAP1 in Tumor 4 (Fig. 2a). Two reads were obtained for HPV16 in LRP1B of Tumor 5, which confirmed the structure of the inserted HPV16 DNA with the additional 982 bp (Fig. 2c).
For the HPV16 DNA insertion in C5orf67 in Tumor 4, over 100 long-range sequencing reads were obtained covering the viral genome and nearby human sequences (Fig. 2d, Supplementary Fig. 3). Of these, 59 of them covered HPV16 sequences, some including one or both of the two junctions identified by Illumina sequencing, as well as some reads that covered the entire HPV16 DNA insertion plus flanking human DNA on both sides, thus confirming the overall structure and both junctions. Moreover, a 28 kbp segment of the human genome with the HPV16 DNA insertion site near the center had on average about 4- to 5-fold higher coverage than the flanking human DNA (Fig. 2d). This indicated that a segment comprising about 36 kbp of human plus HPV16 DNA (~26 and ~8 kbp, respectively) was amplified relative to nearby sequences on chromosome 5.
Since HPV DNA is sometimes present in episomal (extra-chromosomal, circular) form linked to human DNA in tumors20,28,29, we analyzed the long-range sequence reads at the two boundaries of the amplified, chromosome 5 segment to determine if they were joined (Supplementary Fig. 3). We identified 55 long-range reads in which the reference human genome (hg38) positions 56,528,445 and 56,554,989 were precisely continuous (Fig. 2d, Supplementary Fig. 3). This junction was consistent with either a 36 kbp episome or a concatemer of the identical 36 kbp sequence integrated in the human genome (Fig. 2d). The formation of such an episome could be explained by a multistep process. First the subgenomic segment of HPV16 integrated into one allele of C5orf67. After integration, the two sites in human DNA flanking either side of the viral DNA insertion rearranged and joined together to form a circle (Fig. 2d, Supplementary Fig. 3). Presumably, the HPV-human hetero-chimeric episome then attained an average 4- to 5-fold level of amplification by DNA replication processes, possibly involving HPV-specific replication mechanisms20,28,29. However, the sequencing data do not exclude the possibility that the 36 kbp hetero-catemeric segment is oligomerized as tandem, 36 kbp, direct repeats in a larger episome, nor the possibility that the 36 kbp hetero-catemers might be present as tandem repeats that are integrated with the human genome (Fig. 2d), or even that both episomes and integrated tandem repeats might present in the tumor. Hetero-catemers may occur frequently in HPV-induced tumors20,28,29,30. While it can be argued that episomal hetero-catemers are not integrated into the human genome per se, the HPV segments in them are each joined by two junctions with the human genome just like an integration event in a human chromosome, and arguably derived from such events. The long-range sequencing additionally showed that the normal, “empty” allele of C5orf67 without the HPV16 DNA insertion was also present in the tumor. This analysis emphasizes the strength of the HC + SEQ and long-range sequencing approaches to elucidate the detailed, large-scale structures of integrated HPV DNAs including human-HPV16 DNA hetero-catemeric structures.
To summarize, Tumors 2, 4, and 5 exhibited the presence of integrated HPV16 DNAs. In Tumors 2 and 4, subgenomic segments of HPV 16 DNA were identified, while in Tumor 5, a tandem array comprising at least one full-length HPV16 genome was observed. The insertion in Tumor 4 was a hetero-concatemeric, 36 kbp episome and/or an integrated tandem repeat that was chimeric with human DNA. Two of the three inserted viral DNAs were flanked by short direct repeats. The viral DNAs all included an intact segment with the URR regulatory sequences upstream of the E6 and E7 genes as appropriate for transcription from the viral early promoter, emphasizing the importance of these viral genome components for tumorigenesis. In contrast, the late genes were situated upstream of the URR and HPV transcription start sites, and thus improperly positioned to be transcribed from the viral URR or late (in E7) promoters. Such an overall structure is characteristic of HPV DNA integrations in other HPV induced tumors21,31, and presumably is important for expression of the viral E6 and E7 oncogenes.
While co-infections were detected in both Tumors 10 and 13, no human-viral DNA junction sequence reads were detected. Viral DNA presence was confirmed by PCR in both tumors using primers designed for E6 or E7 components of the specific virus (Supplementary Fig. 4). In Tumor 10, there was full coverage for both HPV6 and HPV16. Oddly, reads were detected crossing an intraviral DNA junction between nucleotides 1807 (within E1) and 7135 (within L1) of HPV16 (Fig. 1). The segment extending from L1 through the URR, E6, E7 and into E1 had about 40% as many reads as rest of viral genome, suggesting that, in addition to full-length HPV16, an unusual, unintegrated, subgenomic segment from 1807 through 7135 was somehow present in at least a fraction of the cells in the tumor. Tumor 13 showed the full genome of HPV62, while the HPV53 genome had the intraviral genome with a deletion from nucleotides 4825 to 5297 (Fig. 1). The presence of deleted forms of HPV in the tumors at various levels is consistent with the ideas that the viral DNA is unstable in tumors, and the absence of junctions between any of the HPVs in Tumors 10 and 13 with human DNA might suggest that these viral DNAs were episomal rather than integrated into the human genome, or perhaps that the sampled portion of the tumor lacked integrated DNA.
In vulvar tumors with integrated HPV DNA, viral RNA transcripts are limited to the early genes and predominantly encode full-length E7 and the E6*I spliced isoform open reading framesBased on RNA-sequencing (RNA-seq) analyses of bulk tumor tissue, viral transcripts were found to encompass only a fraction of the HPV transcriptome. RNA-seq reads were aligned to their respective HPV type reference genomes from PAVE10 using STAR v2.7.9a25. Coverage patterns (Fig. 3) in each of these tumors reflected mRNA transcripts derived from the early promoter, encompassing E6, E7, and the very beginning of E1, with viral genome coverage extending precisely to the positions where viral DNA was joined to human DNA. There was extensive use of viral splice junctions. Notably, almost all transcripts from the E6 gene had the E6*I intron excised in all three tumors (Fig. 3). This small intron spans 182 nucleotides from positions 227 to 408 within the E6 ORF. Excision of it results in mRNAs encoding a frameshift at the splice junction that results in a C-terminally truncated form of E6 protein. The tumors all contained levels of full-length E6 open reading frame (ORF)-containing mRNAs that were far lower than those for the E6*I and E7 ORFs. There was also extensive use of the 5’ splice site (5’ss) in E1 in all three tumors, and 3’ splice sites (3’ss) for E4 in Tumors 2 and 4. For Tumor 5, the only viral transcripts detected downstream of the E1 5’ss were a low level of transcripts from the E1 region, with no transcripts from E2, E4, and E5 identified, as expected from the integrated DNA structure. Likewise, no transcripts from the disrupted E5 region were detected in Tumor 2. Tumor 4 did contain an intact E5 ORF in the inserted HPV16 DNA (Fig. 2). However, part of the 3’ untranslated region was truncated by the insertion junction at position 4136 situated 35 bp downstream of the E5 ORF stop codon and upstream of the canonical AATAAA polyadenylation signal, and thus, predictably, only few E5 transcripts were detected in that tumor. The quality of RNAs obtained from HPV DNA-containing Tumors 10 and 13 was too low to yield RNA-seq data.
Fig. 3: RNAseq analysis of HPV transcripts in three vulvar tumors harboring integrated HPV16 DNA.
The plots depict read counts aligned to the HPV16 genome using IGV, with the Y-axis representing the scale of read counts for each plot, and read counts displayed as counts per million reads. At the bottom, the full-length HPV16 genome is shown, indicating the positions of the viral open reading frames (ORFs) represented as boxes. The large blue arrow indicates the 5’ to 3’ transcriptional orientation of all the viral ORFs. The standard numbering scale for the HPV16 genome is shown at the top. The genome is linearized between the early (E) and late (L) ORFs to position the URR-E6-E7 segment centrally as is characteristic of integrated HPV genomes in tumors (see Fig. 2) including the URR immediately upstream of the transcribed E6 and E7 ORFs. The viral nucleotide immediately preceding the junction with human DNA is numbered at the end of each plot. HPV RNA 5′ and 3′ splice sites (5′ss and 3′ss) are labeled and indicated by the vertical dashed lines. These include the E6*I splice sites, responsible for a frame-shifting deletion of much of the E6 coding segment, and the 5′ss (E1^E4 5’ss) just downstream of the E1 start codon that typically splices to the 3’ss for the E4 ORF, and also can join to 3’ splice sites in the human genome downstream of inserted HPV DNAs. No transcripts from the viral L regions were detected in any of the tumors.
Transcription from each of the three integrated HPV DNAs extended from viral sequences into the human sequences (Fig. 4a–c). In all three instances, the transcribed human segment derived from the antisense strand of the intron of the human gene in which the viral DNA was inserted as the template, with the length of the human portion of the transcripts varying among the three insertions. In some instances, the transcripts comprising the sequences immediately downstream of the inserted viral DNA were sufficiently stable to be detected. In others, the human sequences were joined by presumably cryptic 3’ss’s to the upstream viral RNA by a splice junction, principally the E1^E4 5’ss (that splice site is indicated in Fig. 4a–c). The human portions of such transcripts were antisense to intronic sequences of the human genes in which the viral DNAs were integrated. Levels of spliced mRNAs (the positive sense transcripts) for each of the three human genes were assessed in the RNA-seq dataset (Fig. 4d–f). Transcripts were quantified and normalized for total read counts using Salmon32 prior to differential gene expression (DGE) analysis. All three human genes were expressed at only relatively low levels with NCKAP1 the highest of the three, suggesting that the HPV URR enhancer had no substantive effects on expression of these human genes.
Fig. 4: Transcription of human genes with HPV16 DNA insertions determined from RNAseq data.
a, b, c Positions of human RNA sequences derived from the antisense strand of the cognate human genes (brown) fused with HPV16 RNA sequences (blue) for each tumor. The top line in each displays the chromosomal position and 5′ to 3′ transcriptional orientation of the human gene. Below that, IGV plots show the exon-intron structure of human gene, with the HPV16 DNA insert displayed in a lighter shade of blue. The boxed regions within each gene highlight the segments containing HPV16 DNA, with spliced fusion transcripts shown below the DNA for detailed visualization of viral genetic detail. The scales of the HPV16 DNA are larger than the flanking human DNA segments to allow transcriptional features to be presented. The brown arcs represent the introns excised from HPV16 5’ splice sites (5’ss) to presumably cryptic 3’ splice sites (3′ss) of the human genes. The numbers within the arcs indicate the RNAseq read counts for each splice site junction. The majority of the transcripts derived from HPV16 exhibited splicing at the major E6* splice junction known as E6*I (blue arcs), with the positions of the junctions at nucleotides 226 and 409 shown in blue. Most splicing from HPV sequences to human gene antisense sequences involved the 5’ss at position 880 at the beginning of the viral E1 gene open reading frame as shown. For Tumor 5 (c), a smaller number of spliced RNAs alternatively involved the 5’ss at position 226 within the E6 gene, which joined a 3′ss of LRP1B. In Tumor 4, numerous transcripts were spliced from HPV16 sequences originating at the viral late promoter and entailed the 5’ss at position 1302 labeled E2M. d, e and f show the total numbers of RNAseq reads derived from the sense strands of the indicated human genes, aggregated for each gene. The total deduplicated read counts (counts per million reads) from each human gene are plotted separately for the HPV-positive and the HPV-negative vulvar cancers. Specifically, for tumors 1–9. Each panel utilizes a distinct Y-axis read count scale for the corresponding sample. The black arrows indicate samples in which HPV16 DNA was integrated into the specific human gene.
Differential expression analysis of global human gene transcription between HPV-positive and negative VSCCsTo investigate whether the global transcriptome of HPV-positive vulva cancers differed from that of HPV-negative ones, human genome-wide transcriptomic analysis was performed on tumors with an RNA integrity number (RIN) above 6. The normalized sequencing read counts of nine tumors, three HPV positive (Tumors 2, 4, 5) and six HPV negative (Tumors 1, 3, 6, 7, 8, 9), were subjected to DGE analysis. Due to the small sample size, and to prevent one sample from dominating the differential results, we required at least three samples in any comparative group to have ≥10 normalized counts for any individual gene. Following prefiltering of genes with no counts, we found 16,532 genes out of 37,951 total normalized genes to fit these criteria. First to compare the HPV-positive and negative samples, principal component analysis (PCA) was performed on the regularized, logarithm-transformed counts of RNA-seq reads33. PC1 and PC2 accounted for 46.5% of the variability. HPV status was substantially represented in the first principal component (Supplementary Fig. 5A-C). Additional biological covariates assessed included whether the tumor was primary or recurrent disease, and if the patient received prior chemotherapy or radiation therapy. These covariates were represented primarily in PC5, and both were subsequently included in the linear model for DGE analysis. Hierarchical clustering of the 50 most variable genes showed that Tumors 2 and 4, two primary HPV-positive tumors, clustered together (Supplementary Fig. 5D). Curiously, over half of the 50 most variably expressed genes identified are involved in adaptive immune system function including human leukocyte antigen (HLA) genes and immunoglobulin genes.
To analyze differential gene expression patterns of HPV positive versus negative tumors further, we set a false discovery rate (FDR) of <0.1 and found 402 genes (2.4%) to be upregulated and 327 (2%) to be downregulated in HPV-positive tumors compared to HPV-negative tumors (Supplementary Fig. 5E and Supplementary Table 2). The top 10 differentially expressed genes by adjusted p-value were MSH5, STAG3, RNF212, SYCP2, MBOAT7, TBC1D3, PORCN, DHX16, ZFR2, and DES (Supplementary Fig. 6). Interestingly, four of these genes (STAG3, RNF212, SYCP2, ZFR2) were previously observed to be differentially expressed in other types of HPV-induced cancers34,35,36,37, indicating similarities among these virally-induced tumors. GO categorization and over-representation indicates that in addition to the significant enrichment of cell surface proteins essential for the adaptive immune system, HPV-positive vulvar tumors were associated with the activation of programs of ECM remodeling as reported for other HPV associated tumors (Supplementary Fig. 7)38,39,40.
Because several markers pointing to adaptive immune response emerged as the most variable genes overrepresented in the HPV positive samples, we applied the cell subtype deconvolution algorithm CIBERSORTx41 to estimate the relative proportions of immune cell subtypes in the nine vulvar carcinoma samples based on bulk tumor gene expression profiling. Despite the inherent variability attributed to the small sample size, our analysis revealed intriguing findings in relation to the immune subtypes. Notably, hierarchical clustering analysis demonstrated distinct immune gene signatures between HPV-positive and HPV-negative tumors. This observation implies that, despite variations in immune subtype abundances, HPV-positive and HPV-negative tumors possess discernable functional properties that characterize them, thereby providing valuable insights into the underlying biological mechanisms involved (Fig. 5a). In HPV-positive Tumors 2 and 4, there was a relative enrichment of plasma cells, albeit not at statistical significance (Wilcoxon p = 0.06) (Fig. 5b). Tumor 5, which was also HPV-positive, did not show this enrichment, however this sample was taken from a disease recurrence site, and in addition, the patient had received both prior chemotherapy and radiation therapy, unlike Tumors 2 and 4 which were primary disease tissue biopsies from treatment naïve individuals. An additional pattern noted was that several HPV-negative tumors (Tumors 1, 3, 6, 7, 8) exhibited relative enrichment of non-activated M0 macrophages (Wilcoxon p = 0.02) (Fig. 5c). EPIC42 was also used to estimate tumor infiltrating cells in the HPV-positive versus negative tumors including stromal cells and endothelial cells in addition to condensed sets of immune cells (Fig. 5d, e). While the HPV-positive and HPV-negative vulvar cancers yielded similar infiltrating cell profiles, some differences were noted. The endothelial cell fraction was significantly higher in the HPV-positive compared to the HPV-negative tumors (p = 0.04). Furthermore, it was observed that the HPV-positive group of Tumors 2, 4 and 5 exhibited high overall levels of B cells. Although this did not reach statistical significance, perhaps it reflected the elevated plasma cell subsets observed in Tumors 2 and 4 with CIBERSORTx (Fig. 5a). Although statistical significance was not achieved, this finding is concordant with the observations obtained using CIBERSORTx. This finding may provide insight into the potential relationship between elevated B cell levels and the presence of plasma cell subsets in these HPV-positive tumors.
Fig. 5: Evaluation of tumor infiltrating immune cells using RNAseq expression data.
a The relative frequencies of 22 immune cell gene expression profiles across nine tumor samples were assessed using CIBERSORTx. HPV-positive tumors, identified through hybridization capture analysis, are labeled in red, while HPV negative tumors are labeled in black. On top of the figure a color-coded key to relative values. Tumors 2 and 4, both HPV-positive, exhibited relatively higher proportions of plasma cells in the tumor microenvironment. In addition, all three HPV-positive tumors displayed lower relative levels of M0 macrophages compared to HPV-negative tumors. EPIC was employed to further examine the tumor microenvironment and provide a condensed representation of immune cell populations in the HPV-positive and HPV-negative samples. Differences in plasma cells (b) and M0 macrophages (c) were observed. The tumor microenvironment was further evaluated using EPIC to provide a condensed set of immune cell populations in addition to cancer-associated fibroblasts and endothelial cells between HPV-positive (d) and HPV-negative tumors (e). Differences in (b) cells as a population whole did not meet statistical significance. However endothelial cells were noted to be more prevalent in HPV positive tumor’s TME compared to HPV-negative tumors (p = 0.04).
Comparison of HPV-positive and HPV-negative vulvar cancers by whole genome sequencing (WGS)Molecular genetic information about vulvar cancers is limited. In addition to the insights provided here about HPV-positive tumors, it was of particular interest to examine HPV-negative vulvar cancers and compare them to HPV-positive tumors. Therefore, we undertook whole genome, short-range, Illumina sequencing on all 13 tumor samples in this study. WGS analysis of the HPV positive (n = 5) and negative (n
留言 (0)