Study population and samples
The study cohort consisted of 110 patients with pathologically confirmed gastric cancer and 139 participants with non-cancerous conditions enrolled at Zhejiang Cancer Hospital (Hangzhou, China) between October 1st, 2021, and May 30th, 2022. Following the completion of training cohort enrollment, a separate validation cohort comprising 73 gastric cancer patients and 94 non-cancer individuals was collected at the same site from June 1st, 2022, to October 31st, 2022 (Additional file 1: Fig. S1). All participants provided written informed consent. Each participant had a peripheral blood sample (~ 10 ml) collected before other screening tests for gastric cancer, including gastroscopy, CT scans, and blood tests of tumor markers. Plasma was isolated from blood samples within 4 h. cfDNA was then extracted within 72 h and stored at – 80 ℃ for further cfDNA sequencing. All processes were conducted in Zhejiang Cancer Hospital (Hangzhou, China). Both cohorts followed the same protocols for participant enrollment and sample preparations, except that 31 samples collected between September 2022 and October 2022 were processed with a new automated liquid handling platform.
A second validation cohort consisting of cfDNA samples of 47 gastric cancer patients and 49 non-cancer individuals was retrospectively collected from an independent center, Ningbo No.2 Hospital (Ningbo, China). Cases in the Biobank were first filtered using the same inclusion and exclusion criteria as the study cohort. Stratified random selection was then performed to ensure that the demographic distributions (age, sex, postsurgical stage, tumor location, Lauren’s classification, and non-cancerous disease status) of this validation cohort mirrored those of the study cohort. Sample preparations followed the same protocols as the study cohort.
The study was approved by the institutional review board of Zhejiang Cancer Hospital and performed in accordance with the principles of the Declaration of Helsinki.
Cohort inclusion and exclusion criteria
Participants meeting the following criteria were considered eligible for enrollment: (1) individuals aged between 18 and 85 years old; (2) individuals without a prior history of cancer; (3) individuals with a plasma sample that was collected before initial screening tests and passed QC; and (4) individuals who provided informed consent. Eligible individuals were assigned to the appropriate group if they further met the following criteria: (1) gastric cancer group: individuals who were pathologically confirmed with gastric adenocarcinomas by biopsies without concomitant malignancies and received curative surgeries for gastric cancer; and (2) non-cancer group: individuals who showed no signs of cancers based on routine physical examinations, including blood tests, ultrasound and CT, and no sign of gastric cancer from gastric cancer-specific screening tests. The size of the non-cancer group was controlled to match the gastric cancer group.
Participants meeting any of the following criteria were excluded from the gastric cancer group: (1) individuals who were pathologically diagnosed with stage III/IV gastric cancer; (2) individuals who withdrew informed consent; and (3) individuals whose sequencing data failed QC.
Participants meeting any of the following criteria were excluded from the non-cancer group: (1) individuals with significant chronic diseases of other systems, such as severe cardiovascular diseases, uncontrolled diabetes, hypertension, and infectious diseases; (2) Individuals who had abnormal results of tumor marker (CEA, CA19-9, CA125, PSA, AFP, etc.) examinations within the past year; (3) individuals who withdrew informed consent; and (4) individuals whose sequencing data failed QC.
Sample and sequencing library preparations
Peripheral blood samples were first centrifuged at 16,000 × at 4 °C for 10 min to aliquot plasma and serum separately. Circulating cell-free DNA (cfDNA) was isolated from 2 to 4 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen), following the manufacturer’s protocols. The concentrations of isolated cfDNA were examined using Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). cfDNA libraries for whole-genome sequencing (WGS) were prepared using the KAPA Hyper Prep Kit (KAPA Biosystems) according to the manufacturer’s protocol. In brief, 5–10 ng of cfDNA per sample was subjected to end-repairing, A-tailing, and ligation with adapters sequentially. The Hamilton Microlab STAR automated liquid handling platform (Hamilton Company) (hereafter denoted as Platform 1) was used for the automated pipeline operations. For 31 samples in the first validation cohort that were collected between September 2022 and October 2022, the Beckman Biomek i7 automated liquid handling platform (Beckman) (hereafter denoted as Platform 2) was used. The libraries were quantified using the KAPA SYBR FAST qPCR Master Mix (KAPA Biosystems).
Low-coverage whole-genome sequencing and alignment
Whole-genome libraries were sequenced using 100-bp paired-end runs on the NovaSeq platforms (Illumina) at 8X coverage per genome according to the manufacturer’s instructions. Trimmomatic [7] was used for FASTQ file quality control. The Picard toolkit was used to remove PCR duplicates (http://broadinstitute.github.io/picard/). Qualified reads were then aligned to the human reference genome (GRCh37/UCSC hg19) using the Burrows–Wheeler Aligner (BWA) [8]. To mitigate potential biases introduced by varying sequencing depths among samples, all plasma samples were down-sampled to a uniform coverage of 5 × .
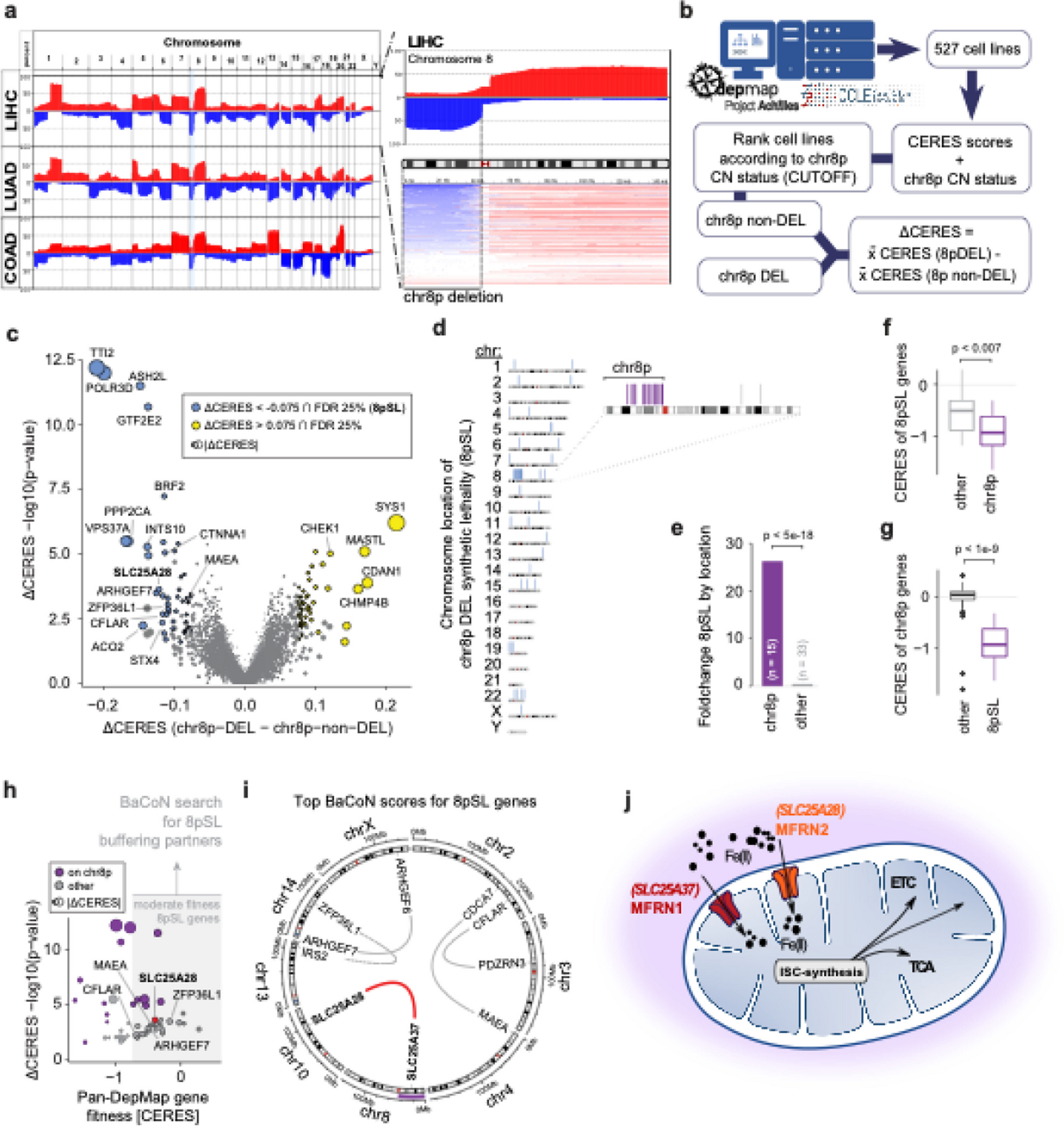
Whole-genome sequencing featuresFragment size pattern
The extraction of fragment size features was adapted from the method introduced by D. Mathios et al. [9]. A non-parametric method for fragment-level adjustment for GC content and library size was first performed. We tiled the reference genome into non-overlapping 5-Mb bins and preserved a total of 541 bins with an average GC content ≥ 0.3 and an average mappability ≥ 0.9. We then computed the adjusted number of short (100–150 bp) and long (151–220 bp) fragments in each bin and standardized the numbers to have mean zero and unit standard deviation across all bins.
Copy number variation
The ichorCNA tool was used to profile copy number variations as described by Wan et al. [10]. The reference genome was tiled into non-overlapping 1-Mb bins, totaling 2475 bins. The depth of each bin was used to compare against the software baseline and compute the log2 ratio.
Nucleosome coverage pattern
The cfDNA fragmentation coverage patterns at transcription start sites (TSSs) were reported to be associated with epigenetic regulation. Therefore, cfDNA coverage patterns at certain TSS regions may aid the detection of cancer. The selection of TSSs and profiling of related nucleosome coverage patterns followed the method described by Doebley et al. [4]. We used the GTRD database (v 21.12, https://gtrd.biouml.org/downloads/21.12/chip-seq/) [11] to select transcription factors (TFs) with more than 10,000 highly mappable sites. TFs falling beyond a list of TFs with known binding sites in the CIS-BP database (v2.00, http://cisbp.ccbr.utoronto.ca/bulk.php) [12] were excluded. A total of 854 TFs were finally selected. For each TF, we selected 10,000 mappable sites with the highest peak counts for the analyses of coverage patterns.
To profile the nucleosome coverage pattern of a TF, we first extracted all reads of lengths between 100 and 220 bp in a window (− 5 kbp to + 5 kbp) around each binding site of the TF. For each read, we assigned a weight of the reciprocal of its GC bias. For each site, we split it into 15-bp bins and summed the weights of all reads whose midpoints fall within each bin, generating a GC-corrected midpoint coverage profile. For each TF, we averaged the midpoint coverage profiles of all its binding sites and normalized the 10-kbp window coverages to a mean of 1. The resulting coverage curve along the 10 kb window was then smoothed using a Savitzky-Golay filter with a window length of 150 bp and a polynomial order of 3 to generate the final coverage profile of the TF.
We extracted three features to quantify the coverage profile of each of the 854 selected TFs: (1) the mean coverage of the region flanking 1 kbp upstream and 1 kbp downstream of the center; (2) the coverage at the center; and (3) the amplitude of coverage peaks surrounding the center by a fast Fourier transform.
Single nucleotide substitution signature
The single nucleotide substitution (SNS) signatures were derived from six types: C > A, C > G, C > T, T > A, T > C, and T > G. Taking into account one upstream and one downstream adjacent base, a total of 6 × 4 × 4 = 96 combinations were involved. The relative abundance of each combination was calculated and denoted as the signature. The processes were adapted from the method developed by Wan et al. [6].
Raw FASTQ files were first trimmed and aligned to the hg19 genome, and duplicate reads were removed before sorting and indexing into BAM files by SAMtools (version 1.9) [13]. The average sequencing depth was calculated from the BAM files. Reads of alternative alignment or template length > 300 bp were excluded. Repeat or low-complexity regions were masked before further analyses. GC bias metrics were calculated using Picard (version 2.19.0, http://broadinstitute.github.io/picard/) with a bin size of 400 bp, and the average GC profile, which were later used to normalize the mutation counts by the GC content of each read, were determined by LOESS smoothing method. Paired reads with only 1 mismatch with reference genome were retained, and BCFtools (version 1.9) [14] mpileup were used along with multiallelic-caller to call SNS mutations from reads that met the following criteria: base quality ≥ 30 and mapping quality ≥ 60. InDels were not included. The identified mutations were annotated by ANNOVAR (version 2016–04-25) using RefSeq and dbSNP [15]. SNP sites were then filtered out from the SNS variants to minimize the influence of germline mutations. Clonal hematopoiesis of indeterminate potential (CHIP) mutations was filtered out using an in-house list of frequent CHIP mutations generated from a normal pool of 1000 healthy individuals. The filtered SNS variants were categorized into the 96 types as described above. Counts of each type were calculated and normalized based on the mean sequencing depth.
To further reduce the effects of potential noises from various sources in healthy populations, a baseline control was built from an internal sample pool of 300 healthy individuals. The 96-SNS signatures of each healthy control were generated through the same processes as described above. The baseline was set as the mean value over the 300 controls for each SNS signature. The final SNS signatures of a test sample were calculated as the raw signature values subtracting the baseline values.
Machine learning and cross-validation analyses
We built a two-layer machine learning classifier for the malignant nodules and benign ones. The first-layer module took one type of the features (FSP, NCP, CNV, SNS) as the input. For each feature type, the module iterated through four algorithms, elastic-net logistic regression (GLM), extreme gradient boosting (XGBoost), random forest (RF), and neural network (NN). Hyperparameter tuning was conducted using random search across a list of candidate values for each algorithm. These processes resulted in the generation of over 200 models. From this pool, we identified five algorithm-hyperparameter combinations that yielded the highest classification area under the receiver operator characteristic curve (AUROC) metrics, without consideration for the underlying algorithms. These top-performing combinations were selected as the first-layer 20 base models (4 feature types X 5 best algorithm-hyperparameter combinations = 20 base models) for later second-layer stacking.
The second-layer module stacked the 20 base models with algorithm iteration (GLM, XGBoost, and RF) and hyperparameter tuning, also generating over 100 candidate algorithm-hyperparameter combinations. From these combinations, five stacked models with the best AUROC metrics were saved. The prediction scores from these five models were averaged to generate the final outputs of the classifier. In the study cohort, the classifier was evaluated through fivefold cross-validations. For the validation cohort, the models were fitted using the full study cohort and then used to predict the validation samples.
Rank-based variable importance
The default variable importance from different machine learning algorithms had different magnitudes and thus could not be merged. Our assay stacked models of multiple algorithms for each feature type. To evaluate the relative importance of feature variables across these models, we used their ranks in the default variable importance in each base model, which were of no magnitude. For each base model, we assigned a rank score for 25 variables of the highest default importance from 25 to 1. All variables ranked beyond 25th would be assigned a uniform score of 1. Each base model was assigned a weight score based on its rank in the second-layer model. For the ensemble model, 20 base models were stacked. The weight scores thus ranged from 20 for the most important base model to 1 for the least. The rank scores of a variable in different base models were then averaged with the weights. Within each feature type, the relative rank-based importance of a variable was calculated as its weighted average rank score, standardized to the max score of this feature type. This ensures that the most important variable has a relative importance value of 1.
Simulation for estimating assay performance in a screening population
To assess and compare the performance of gastroscopy and our cfDNA-based assay in a hypothetical screening population of 100,000 individuals, we used Monte Carlo simulations to capture the uncertainty of parameters such as the sensitivity, specificity, participant compliance rate, and gastric cancer prevalence. These parameters were sampled with prior distributions centered on empirical estimates obtained from published large-scale studies and this study. The method was adapted from a published study by Mathios et al. [16].
Hamashima et al. reported a sensitivity of 88.6% (95% CI: 69.8–97.6%) and a specificity of 85.1% (95% CI: 84.3–85.9%) for gastroscopy from 7388 screenings [17]. With the R package epiR (version 1.0–14, https://fvas.unimelb.edu.au/research/groups/veterinary-epidemiology-melbourne), we assumed that the prior distributions for the sensitivity and specificity of gastroscopy were Beta(22,3.7) and Beta(6000,1051), respectively. From the performance metrics of our cfDNA-based assay in the validation cohort (sensitivity: 91.8%, 95% CI: 83.2–96.2%; specificity: 94.5%, 95% CI: 84.1–95.6%), we assumed the respective prior distributions as Beta(67,6) and Beta(86,8).
Zeng et al. reported a populational compliance rate of 43.8% for gastroscopic screenings from a multi-center trial of 230,583 subjects [18]. Therefore, the prior model for gastroscopy compliance rate was Beta(43.8, 56.2). For non-invasive cfDNA-based tests, the compliance rate was conservatively estimated at 80.0%, leading to an assumed prior distribution of Beta(80.0, 20.0). The study also provided a populational gastric cancer prevalence of 0.8%. Thus, for a given screening population (N), the number of gastric cancer cases may be estimated with a distribution of Binomial(0.8%, N).
With all these priors, we conducted the Monte Carlo simulation as follows:
1.
Sampled the probabilities of compliance rates (C) from the prior beta distributions described above:
Cendo ~ Beta(43.8, 56.2)
CctDNA ~ Beta(80, 20)
2.
Simulated the number of individuals that completed the tests (N) in the 100,000 screening cohort from binomial distributions with the probabilities of compliance rates (C):
Nendo ~ Binomial(Cendo, 100,000)
NctDNA ~ Binomial(CctDNA, 100,000)
3.
Simulated the number of gastric cancer cases (S) in the participants from binomial distributions with the populational gastric cancer prevalence (0.8%):
Sendo ~ Binomial(0.008, Nendo)
SctDNA ~ Binomial(0.008, NctDNA)
4.
Sampled the sensitivity (SE) and specificity (SP) from the prior beta distributions described above:
SEendo ~ Beta(22, 3.7)
SEctDNA ~ Beta(67, 6)
SPendo ~ Beta(6000, 1051)
SPctDNA ~ Beta(86, 8)
5.
Simulated the number of true positive (TP) and true negative (TP) incidences from binomial distributions:
TPendo ~ Binomial(SEendo, Sendo)
TPctDNA ~ Binomial(SEctDNA, SctDNA)
TNendo ~ Binomial(SPendo, Nendo − Sendo)
TNctDNA ~ Binomial(SPctDNA, NctDNA − SctDNA)
6.
With true positive incidences (TP), true negative incidences (TN), total gastric cancer cases (S), and total testing cases (N), we were able to calculate the negative prediction value NPV = TN/(TN + S − TP) and false negative rate FNR = (S − TP)/N.
We ran the simulation for 10,000 iterations and obtained the posterior distributions of the performance metrics, which allowed for comparisons between the two methodologies in a large hypothetical population with stochastic effects considered.
Gastric tumor location
The locations of gastric tumors were divided into three groups: proximal, middle, and distal. The proximal group included the cardia and fundus zones. The middle group included the stomach body regions. The distal group included the incisura angularis and pyloric antrum zones.
Outcomes and timelines
This observatory study was registered on ClinicalTrials.gov under the identifier NCT05269056 on March 7, 2022. The primary outcome measure of the registered trial is the AUROC metric for differentiating stage I/II gastric cancer patients and non-cancer individuals using the cfDNA-based assay. The secondary outcome measures are the sensitivity and specificity of the assay.
The enrollment dates of the first and last participants in the study cohort were October 1st, 2021, and May 30th, 2022, respectively. The enrollment dates of the first and last participants in the first validation cohort were June 3rd, 2022, and October 31st, 2022, respectively. The model construction and assessment of results in the study cohort started on July 1st, 2022, after the completion of data collection of the study cohort. The assessment of results in the validation cohort started on December 1st, 2022.
Statistical analysis
The comparison of continuous numeric data was done using the Wilcoxon test. The comparison of proportions between groups was done using Fisher’s exact test. The trend of continuous numeric data across ordered groups was assessed using the Jonckheere trend test. A two-sided P value of less than 0.05 was considered significant for all tests unless otherwise indicated. All statistical analyses were performed in R (v4.0.2).


留言 (0)