
記住我
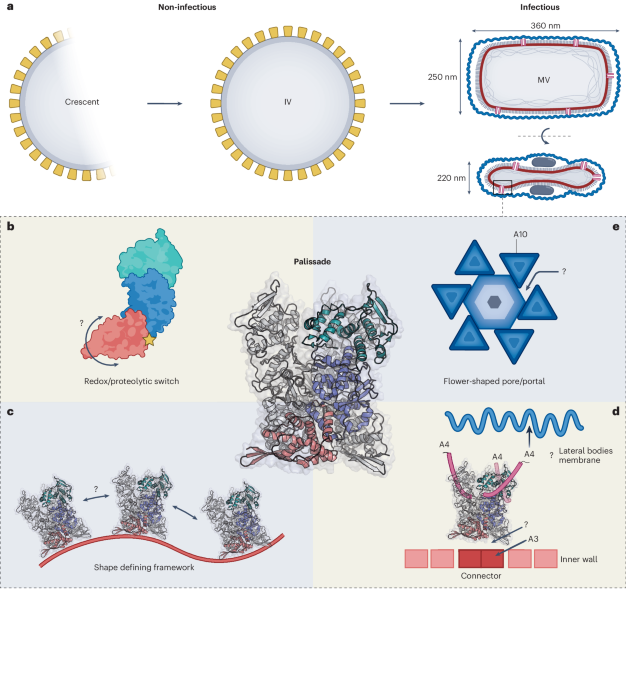
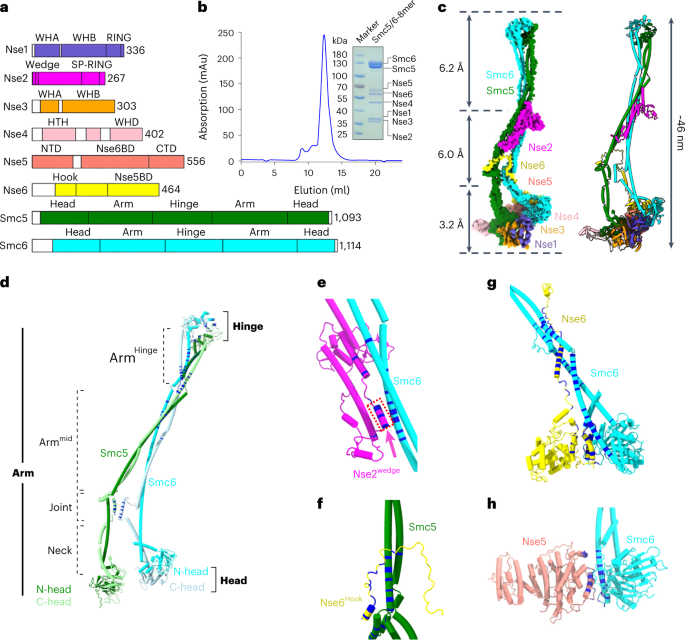
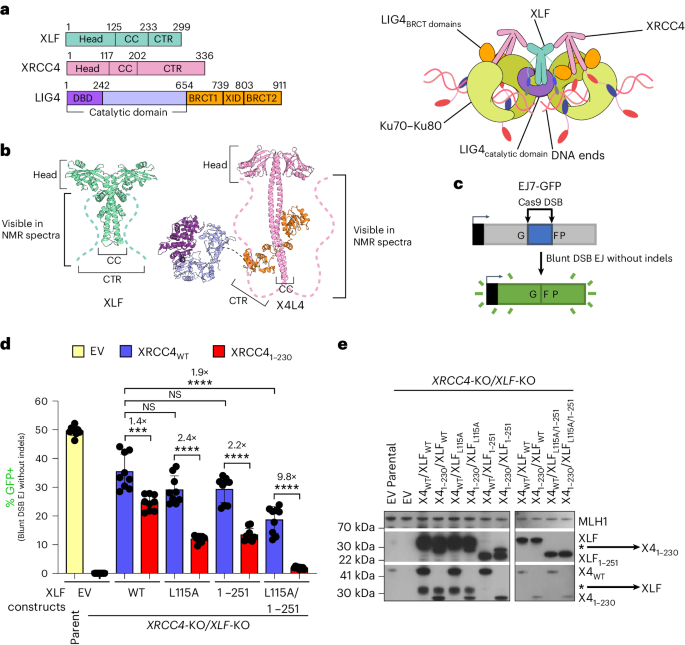
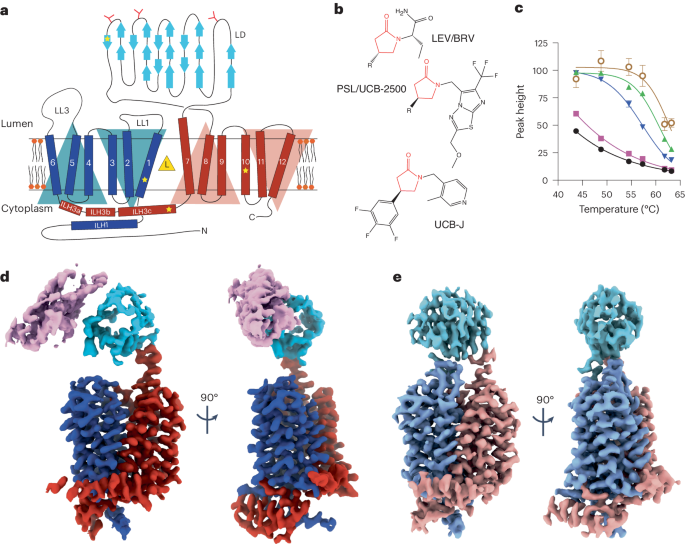

Experiments were carried out using female H9 hES cells obtained from the WiCell Research Institute. Research on hES cells was approved by the Agence de la Biomédecine and informed consent was obtained from all subjects.
Primed H9 hES cells were cultured on Matrigel-coated culture dishes in mTeSR1 medium (StemCell Technologies) according to the manufacturer’s instructions in 5% O2 and 5% CO2 at 37 °C. They were routinely passaged in clumps using gentle cell dissociation reagent (StemCell Technologies) according to the manufacturer’s instructions. For experiments requiring a single-cell suspension, cells were incubated with Accutase (StemCell Technologies) and plated in fresh mTeSR1 medium supplemented with 10 μM Y-27632 (StemCell Technologies).
Naive H9 hES cells were generated by chemical resetting of the H9 primed hES cells using the NaiveCult (StemCell Technologies) or PXGL protocol, as previously described, and cultured on inactivated mouse embryonic fibroblasts (MEFs)23,24. PXGL naive hES cells were cultured in PXGL medium consisting of a 1:1 mixture of DMEM/F12 (Sigma-Aldrich) and Neurobasal medium supplemented with 0.5% N-2 supplement, 1% B-27 supplement, 2 mM l-glutamine, 100 µM β-mercaptoethanol and 1× penicillin–streptomycin (all from Gibco, Thermo Fisher Scientific), as well as 1 μM PD0325901 (Axon Medchem, 1408; CAS: 391210-10-9), 2 μM XAV939 (Cell Guidance Systems, SM38-10; CAS: 284028-89-3), 2 μM Gö6983 (Tocris, 2285; CAS: 133053-19-7), 10 ng ml−1 human leukemia inhibitory factor (LIF; Peprotech, 300-05). Naive hES cells were routinely passaged as single cells every 3 days at a ratio of 1:3 using 1× TrypLE Express (Gibco, Thermo Fisher Scientific) and plated in fresh medium supplemented with 10 μM Y-27632 (StemCell Technologies).
Human preimplantation embryosThe use of human embryos donated to research as surplus of in vitro fertilization (IVF) treatment was allowed by the French embryo research oversight committee (Agence de la Biomédecine) under approval number RE18-010R. Embryos used were initially created in the context of an assisted reproductive cycle with a clear reproductive aim and then voluntarily donated for research once the patients had fulfilled their reproductive needs or tested positive for the presence of monogenic diseases. Informed written consent was obtained from both parents of all couples that donated spare embryos following IVF treatment. French legislation does not include the research project in the consent form; the embryos are donated to research, in general. The Agence de la Biomédecine oversight committee rules which project can use which embryos. Donor compensation is forbidden under French law.
All human preimplantation embryos used in this study were obtained from and cultured in the Assisted Reproductive Technology unit of the University Hospital of Nantes, France, which is authorized to collect embryos for research under approval number AG110126AMP of the Agence de la Biomédecine. Molecular analysis of the embryos was performed in compliance with the embryo research oversight committee and the International Society for Stem Cell Research (ISSCR) guidelines44.
Human embryos were thawed following the manufacturer’s instructions (Sydney IVF Thawing Kit for slow freezing, from Cook Medical; RapidWarmCleave or RapidWarmBlast for vitrification, from Vitrolife). Human embryos frozen at the eight-cell stage were loaded in a 12-well dish (Embryoslide, from Vitrolife) with nonsequential culture medium (G-TL, from Vitrolife) under mineral oil (liquid paraffin, from Origio), at 37 °C in 5% O2 and 6% CO2.
Generation of XACT and XIST KO cell lineSingle-guide RNA (sgRNA) sequences flanking the XACT and XIST promoters and first exon were obtained using the web-based tool CRISPOR (http://crispor.tefor.net/) and are provided in Supplementary Table 9. sgRNAs were cloned under a U6 promoter into the pSpCas9(BB)-2A-GFP (green fluorescent protein) (a gift from Feng Zhang; Addgene 48138) and the pSpCas9(BB)-2A-mCherry (generated in house, by replacing GFP with an mCherry reporter using the NEBuilding HiFi DNA Assembly Cloning Kit (New England Biolabs)45. Using the Amaxa 4D-Nucleofector system (Lonza), one million primed H9 hES cells were transfected with 2.5 µg of each plasmid (to a total of 5 µg). Cells were sorted by fluorescence-activated cell sorting (INFLUX 500-BD, BioSciences) 48 h after transfection. Double-positive cells were plated onto a Matrigel-coated 6-cm Petri dish in mTeSR medium supplemented with 1× CloneR (StemCell Technologies). Individual colonies were manually placed into 96-well plates ~10 days after transfection. Deletions and inversion events were screened by PCR. Primer sequences can be found in Supplementary Table 10. XACT and XIST KO H9 primed hES cells were reprogrammed to naive using the NaiveCult and PXGL protocols, respectively, as described above.
CRISPR inhibitionA sgRNA targeting the XIST promoter was designed using the web-based tool CRISPOR (http://crispor.tefor.net/) and cloned into the PB_rtTA_BsmBI vector (a gift from Mauro Calabrese; Addgene 126028)46. The sgRNA sequence can be found in Supplementary Table 9. Using the Amaxa 4D-Nucleofector system (Lonza), one million primed H9 hES cells were transfected with 1.5 µg of PB_tre_dCas9_KRAB (a gift from Mauro Calabrese; Addgene 126030), 0.75 µg of PB_rtTA_BsmBI and 1.5 µg of piggyBac transposase47. Cells were then treated with G418 (350 µg ml−1) and hygromycin (350 µg ml−1) until separate colonies were obtained. The number of random insertions in the genome was verified by qPCR and clones with the lowest insertion number (n = 2) were used for further experiments. For XIST depletion, PXGL-reprogrammed CRIPSRi naive hES cells were treated with 1 µg ml−1 Dox for 10 days.
CUT&RUNCUT&RUN was performed as previously described48. Briefly, 0.3 million cells per replicate were bound to 20 µl of concanavalin A-coated beads (Bangs Laboratories) in binding buffer (20 mM HEPES, 10 mM KCl, 1 mM CaCl2 and 1 mM MnCl2). The beads were washed and resuspended in DIG Wash buffer (20 mM HEPES, 150 mM NaCl, 0.5 mM spermidine and 0.05% digitonin). The primary antibodies (1:50) were added to the bead slurry and rotated at room temperature for 1 h. The beads were washed with DIG Wash buffer and protein A–MNase (micrococcal nuclease) fusion protein (1:400, produced by the Institut Curie Recombinant Protein Platform, 0.785 mg ml−1) was added, before rotating at room temperature for 15 min. After two washes, the beads were resuspended in 150 µl of DIG Wash buffer and the MNase was activated with 2 mM CaCl2, before incubating for 30 min at 0 °C. MNase activity was terminated with 150 µl of 2× Stop buffer (200 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 µg ml−1 RNase A and 40 µg ml−1 glycogen). Cleaved DNA fragments were released by incubating for 20 min at 37 °C, followed by centrifugation for 5 min at 16,000g at 4 °C and collection of the supernatant from the beads on a magnetic rack. The DNA was purified by phenol–chloroform; libraries were prepared using the TruSeq ChIP Library Preparation Kit from Illumina following the manufacturer’s protocol and sequencing was performed on a NovaSeq 6000 instrument (ICGex next-generation sequencing (NGS) platform) to generate 2 × 100 paired-end reads. All the antibodies used in this study are listed in Supplementary Table 11.
RAPseqRAPseq for human XIST was performed as previously described40. The human XIST oligo pool was ordered from GenScript and amplified as previously described to generate single-stranded DNA (ssDNA) biotinylated oligos49. The oligo sequences are listed in Supplementary Table 12. Briefly, 20 million cells were harvested and incubated with 10 ml of freshly made 2 mM DSG at room temperature for 45 min. Cells were further cross-linked with 10 m of 3% formaldehyde for 10 min at 37 °C and the reaction was stopped by adding glycine to a final concentration of 500 mM. Cells were pelleted at 4 °C and stored at 80 °C. Nuclei were isolated in cold lysis buffer (20 mM HEPES pH 7.5, 50 mM KCl, 1.5 mM MnCl2, 1% IGEPAL CA-630 (NP-40), 0.4% sodium deoxycholate and 0.1% N-lauroylsarcosine). Chromatin was solubilized by sonication and fragmented by TURBO DNase digestion (Thermo Fisher Scientific). XIST pulldown was performed on 10 million cells using 1 µg of Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific) and 50 pmol of biotinylated oligos. DNA was eluted by RNase H digestion and cross-linking was reversed by proteinase K digestion at 65 °C for 1 h. Finally, DNA was purified using the GeneJET Gel Extraction kit (Thermo Fisher Scientific); libraries were prepared using the TruSeq ChIP Library Preparation Kit from Illumina and sequencing was performed on a NovaSeq 6000 instrument (ICGex NGS platform) to generate 2 × 100 paired-end reads.
Assay for transposase-accessible chromatin with sequencing (ATAC-seq)ATAC-seq was performed as previously described50. Briefly, 50,000 cells were resuspended in 50 µl of cold lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL CA-630) and centrifuged for 10 min at 500g at 4 °C. The nuclear pellets were resuspended in 50 μl of transposase reaction mix (25 µl of 2× TD buffer, 2.5 µl of transposase and 22.5 µl of H2O) and incubated at 37 °C for 30 min in a ThermoMixer at 1,000 r.p.m. Reactions were cleaned up using the MinElute PCR purification kit (QIAGEN) and DNA was eluted in 10 µl of elution buffer. Transposed DNA was preamplified for five cycles in 50 µl of reaction mix (2.5 μl of 25 μM primer Ad1, 2.5 μl of 25 μM primer Ad2, 25 μl of 2× Master Mix, 10 µl of H2O and 10 μl of transposed elution) with the following cycling conditions: 5 min at 72 °C, 30 s at 98 °C and five cycles of 10 s at 98 °C, 30 s at 63 °C and 1 min at 72 °C. Then, 15 μl of qPCR amplification reaction (5 μl of preamplified sample, 0.5 μl of 25 μM primer Ad1, 0.5 μl of 25 μM primer Ad2, 5 μl of 2× NEBNext Master Mix, 0.24 μl of 25× SYBR Green in DMSO and 3.76 μl of H2O) was carried out with the following cycling conditions: 30 s at 98 °C and 20 cycles of 10 s at 98 °C, 30 s at 63 °C and 1 min at 72 °C. The required number of additional cycles for each sample was calculated. After the final amplification, double-sided bead purification was performed with AMPure XP beads. Final ATAC-seq libraries were eluted in 20 μl of nuclease-free H2O from the beads and were sequenced on a NovaSeq 6000 instrument (Novogene) to generate 2 × 150 paired-end reads.
RNA-seqTotal RNAs were collected using an RNeasy Mini Kit (QIAGEN) and extracted following the manufacturer’s recommendation. Libraries were generated using the Illumina Stranded Total RNA Prep Ligation with the Ribo-Zero Plus kit according to the manufacturer’s recommendation and sequenced on a NovaSeq 6000 instrument (ICGex NGS platform) to generate 2 × 100 paired-end reads.
fastGRO-seqLow-input fastGRO-seq was performed as previously described28. Nuclei were isolated from five million cells and a nuclear run-on assay was performed by adding 25 µl of 2× nuclear run-on buffer (10 mM Tris–HCl pH 8, 5 mM MgCl2, 300 mM KCl, 1 mM DTT, 500 μM ATP, 500 μM GTP, 500 μM 4-thio-UTP, 2 μM CTP, 200 μg ml−1 SUPERase-In and 1% Sarkosyl) at 30 °C for 7 min. RNA was extracted with TRIzol LS reagent (Invitrogen) and quantified using the NanoDrop 2000. A total of 30 µg of RNA was fragmented by sonication and the efficiency was analyzed using the Agilent High-Sensitivity RNA ScreenTape Assay. Fragmented RNA was incubated in biotinylation solution (25 mM HEPES pH 7.4, 10 mM EDTA pH 8.0 and 50 µg of MTS-biotin (Biotium)) for 30 min in the dark at 24 °C and 800 r.p.m. RNA was then precipitated using ethanol and resuspended in nuclease-free water. After DNase treatment, biotinylated RNA was enriched using M280 Streptavidin Dynabeads (Invitrogen) and precipitated using ethanol. Libraries were prepared using the Illumina Stranded Total RNA Prep Ligation with the Ribo-Zero Plus kit according to the manufacturer’s recommendation and sequencing was performed on a NovaSeq 6000 instrument (ICGex NGS platform) to generate 2 × 100 paired-end reads.
MeD-seqMeD-seq assays were essentially performed as previously described51. Briefly, 10 µl of genomic DNA (input 90 ng) from naive hES cells was digested with LpnPI (New England Biolabs) generating 32-bp fragments around the fully methylated recognition site containing a CpG. These short DNA fragments were further processed using the ThruPLEX DNA-seq 96D Kit (Rubicon Genomics). Stem-loop adaptors were blunt-end ligated to repaired input DNA and amplified to include dual-indexed barcodes using a high-fidelity polymerase to generate an indexed Illumina NGS library. The amplified end product was purified on a Pippin HT system with 3% agarose gel cassettes (Sage Science). Multiplexed samples were sequenced on Illumina NextSeq 2000 systems for single-end reads of 50 bp according to the manufacturer’s instructions. Dual-indexed samples were demultiplexed using bcl2fastq software (Illumina).
RNA FISHCell preparation: Primed hES cells were grown on coverslips. Naive hES cells were centrifuged onto Superfrost Plus slides (VWR) using the Cytospin 3 Cytocentrifuge (Shandon). The cells were fixed for 10 min in a 3% paraformaldehyde solution (Electron Microscopy Science) and permeabilized for 5–10 min in ice-cold cytoskeleton (CSK) buffer (10 mM PIPES, 300 mM sucrose, 100 mM NaCl and 3 mM MgCl2 pH 6.8) supplemented with 0.5% Triton X-100 (Sigma-Aldrich) and 2 mM vanadyl ribonucleoside complex (VRC; New England Biolabs).
Probe preparation: RNA FISH probes were obtained after Nick translation of fosmids and BAC constructs purified using phenol–chloroform. Then, 1 μg of purified DNA was labeled for 3 h at 15 °C with fluorescent dUTPs (SpectrumOrange and SpectrumGreen from Abott Molecular and Cy5-UTPs from GE HealthCare Life Science).The templates used in this study were as follows human XIST fosmid (BacPac Resources Center, WI2-3059D20), human POLA1 BAC (BacPac Resources Center, RP11-11104L9), human XACT BAC (BacPac Resources Center, RP11135D3) and human HUWE1 BAC (BacPac Resources Center, RP11-975N19).
Hybridization: First, 100 ng of probes were supplemented with 1 μg of Cot-I DNA (Invitrogen) and 3 μg of Sheared Salmon Sperm DNA (Invitrogen). After precipitation, the probes were resuspended in deionized formamide (Sigma-Aldrich), denatured for 7 min at 75 °C and further incubated for 10 min at 37 °C. Probes were mixed with an equal volume of 2× hybridization buffer (4× SSC, 20% dextran sulfate, 2 mg ml−1 BSA and 2 mM VRC). Coverslips were dehydrated in 80–100% ethanol washes and incubated with the hybridization mix at 37 °C overnight in a humid chamber. Next, the coverslips were washed for 4 min at 42 °C three times with 50% formaldehyde and 2× SSC (pH 7.2) and three times with 2× SSC. The coverslips were mounted in VECTASHIELD PLUS DAPI (Vector Laboratories).
Immunofluorescence stainingPrimed and naive hES cells were prepared as described above and fixed for 10 min in a 3% paraformaldehyde solution (Electron Microscopy Science). Cells were then permeabilized for 7 min with ice-cold PBS supplemented with 0.5% Triton X-100 (Sigma-Aldrich). Cells were blocked in 1× PBS with 1% BSA (Sigma-Aldrich) for 15 min at room temperature and incubated for 45 min at room temperature with primary antibody diluted in 1× PBS with 1% BSA. After three PBS washes, cells were incubated for 40 min at room temperature with Alexa Fluor 488 anti-rabbit or Alexa Fluor 594 anti-mouse secondary antibodies (Life Technologies). Finally, coverslips were mounted in VECTASHIELD PLUS DAPI (Vector Laboratories). For combined Immunofluorescence and RNA FISH, immunofluorescence staining was first performed and then RNA FISH was performed as described above.
Human embryos were fixed at the B3, B4 or B5 stages according to the grading system proposed by Gardner and Schoolcraft52. Embryos were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 5 min at room temperature and washed in 1× PBS with 0.1% BSA. Embryos were permeabilized and blocked in 1× PBS with 0.2% Triton and 10% FBS at room temperature for 60 min. Samples were incubated overnight at 4 °C with primary antibodies. After three PBS washes, embryos were incubated for 2 h at room temperature with Alexa Fluor 488 anti-mouse or Alexa Fluor 568 anti-mouse and Alexa Fluor 647 anti-goat secondary antibodies (Life Technologies) along with DAPI counterstaining (Invitrogen). All primary antibodies used in this study are listed in Supplementary Table 11.
Microscopy and image analysisFluorescent microscopy images for hES cells were taken on a fluorescence DMI-6000 inverted microscope with a motorized stage (Leica), equipped with a charge-coupled device (CCD) HQ2 camera (Roper Scientifics) and an HCX PL APO ×64–100 oil objective (numerical aperture, 1.4; Leica) using the MetaMorph software (version 7.04, Roper Scientifics). Approximately 40 optical z-sections were collected at 0.3-μm steps at different wavelengths depending on the signal (DAPI, 360–470 nm; FITC, 470–525 nm; Cy3, 550–570 nm; Cy5, 647–668 nm). We computed the dispersion of XIST RNA by comparing the cumulative volume of the signal to the theoretical spherical volume it could occupy on the basis of the maximal radial distance. Embryo confocal immunofluorescence images were acquired with an A1-SIM Nikon confocal microscope and a ×20 oil objective. Whole embryos were imaged with 0.5–1-μm optical sections. Stacks were processed using ImageJ and are represented as a two-dimensional ‘maximum projection’ throughout the manuscript.
Total RNA extraction and RT–qPCRTotal RNAs were collected using the RNeasy Mini Kit (QIAGEN) and extracted following the manufacturer’s recommendation. Quantification of the extracted RNAs was performed using the NanoDrop 2000. The DNase step was performed on 1 μg of RNA for 30 min at 37 °C using TURBO DNase (Thermo Fisher Scientific). RNAs were reverse-transcribed using the SuperScript IV kit (Thermo Fisher Scientific) following the manufacturer’s recommendation. Complementary DNAs (cDNAs) were diluted 2.5 times in water and the RNA expression level was assessed by RT–qPCR using the Power SYBR Green Master Mix (Thermo Fisher Scientific) and ViiA-7 Real-Time PCR system (Applied Biosystems). Transcript RNA levels were normalized against the β-actin reference gene following the 2−ΔΔCt method. All RT–qPCR primers used in this study are listed in Supplementary Table 13.
RIPThe cellular extract from 1 million naive H9 hES cells was prepared by adding 1 ml of HNTG buffer (20 mM HEPES pH 7.9, 50 mM NaCl, 1% Triton X-100, 1 mM MgCl2, 1 mM EDTA and 10% glycerol) followed by incubation on ice for 25 min. Cells were then centrifuged at 16,000g at 4 °C for 25 min. The supernatant was collected and 80 µl was used as the input fraction. Next, 30 μl of Magna ChIP Protein G Magnetic Beads (Merck) per immunoprecipitation were washed with PBS–Tween 0.1% three times. Then, 2.5 μg of SPEN antibody (Abcam, AB72266) was added to the beads, followed by 10-min incubation at room temperature with rotation. Beads were washed three times with PBS–Tween 0.1% and resuspended with HNTG buffer. Next, 30 μl of beads were added to the lysate prepared above and incubated for 1.5 h at 4 °C with rotation. After immunoprecipitation, beads were washed three times with 500 μl of HNTG buffer. For RNA preparation, input and immunoprecipitation samples were treated with 200 μg of proteinase K in a total volume of 200 μl for 45 min at 65 °C. RNA was purified using the RNeasy MinElute Cleanup Kit (Qiagen) according to the manufacturer’s recommendations in a final volume of 14 μl of water. The DNase step and RT were performed as described above. The RNA enrichment level was assessed by RT–qPCR using the Power SYBR Green Master Mix (Thermo Fisher Scientific) and ViiA-7 Real-Time PCR system (Applied Biosystems).
siRNA KD SPENSPEN KD was achieved using two different mixes of siRNAs. Mix 1 contained one siRNA (CliniSciences, CRH7929) and mix 2 contained two siRNAs (Thermo Fisher Scientific, 1299001 and 4392420). As a negative control, scramble siRNA was used (ThermoFisher Scientific, AM4611). Naive H9 cells were transfected using the lipofectamine RNAiMAX transfection reagent (ThermoFisher Scientific, 13778030) according to the manufacturer’s recommendations. PXGL medium was changed 1 h before transfection. After 24 h, another round of transfection was performed. After 48 h in total, cells were collected and RNA was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s recommendation.
Raw data processingRNA-seq, fastGRO-seq, CUT&RUN and RAPseq reads were trimmed using trim_galore (version 0.6.5) (https://github.com/FelixKrueger/TrimGalore) with a minimum length of 50 bp. Reads were then mapped to the human genome (hg38) and mouse genome (mm10) using Bowtie 2 (version 2.4.4)53 with the following parameters: ‘--local --very-sensitive-local --no-unal --no-mixed --no-discordant --phred33 -L 10 -X 700’. Reads were then deduplicated using MarkDuplicates from Picard (version 2.23.5) (http://broadinstitute.github.io/picard/) with the following options: ‘--CREATE_INDEX = true --VALIDATION_STRINGENCY = SILENT --REMOVE_DUPLICATES = true --ASSUME_SORTED = true’. BAM files were sorted, filtered (minimum mapping quality = 10) and indexed with SAMtools (version 1.13)54. Reads from MEFs were discarded using the XenofilteR package in R (version 4.1.1)55. Replicate reproducibility was assessed by Pearson’s correlation of the signal using deepTools plotCorrelation.
RNA-seq and fastGRO-seq data analysisFor gene expression analysis, read counts were quantified for each gene with htseq-count from htseq (version 0.13.5)56 using the following options: ‘--stranded reverse -a 10 -t exon -i gene_id -m intersection-nonempty’. The annotation file used was Homo_sapiens.GRCh38.90.gtf. Reads marked by special counters (no feature, ambiguous, too low aQual, not aligned or alignment not unique) were eliminated. For XIST isoform reconstruction, Scallop (version 0.10.4) was used with the default parameters57. XIST isoform quantification was performed using kallisto (version 0.46.2) on the trimmed FASTQ file with the option ‘--rf-stranded’ (ref. 58). For that purpose, the transcriptome FASTA file of exons and introns was generated from the hg38 reference genome using the BUSpaRse package in R (version 4.1.1) and used to produce a kallisto index.
Single-cell expression data analysisFASTQ files from refs. 20,25 were obtained from the ENA (European Nucleotide Archive), under accession numbers PRJEB11202 and PRJNA431392, respectively. STAR (version 2.7.9a) was used for read alignment to the GRCh38 reference human genome. The STAR index was generated using the following options: ‘--runMode genomeGenerate --runThreadN 30 --genomeFastaFiles HS.GRCh38.fa --limitGenomeGenerateRAM 168632718037 --outFileNamePrefix STAR_INDEX. --sjdbGTFfile Homo_sapiens.GRCh38.90.gtf --genomeDir STAR_INDEX_petropoulos_2016 --sjdbOverhang 42’ for Petropoulos data and ‘--genomeDir STAR_INDEX --sjdbOverhang 149’ for Zhou data.
For Petropoulos data, reads (single-end) were aligned using the following options: ‘--soloType SmartSeq --soloFeatures Gene --runThreadN 12 --genomeDir STAR_INDEX_petropoulos_2016 --readFilesCommand zcat --readFilesIn sample_A.fastq.gz --soloStrand Unstranded --soloUMIdedup NoDedup --outFileNamePrefix petro_2016/STAR_ALIGN_sample_A.STARSOLO --outSAMtype BAM SortedByCoordinate --outSAMattrRGline ID:sample_A’.
For Zhou data reads (paired-end) were aligned using the following options: ‘--soloType CB_UMI_Simple --soloCBwhitelist zhou_2019/whitelist.txt --soloFeatures Gene Velocyto --runThreadN 5 --genomeDir STAR_INDEX --readFilesCommand zcat --readFilesIn multiplex_sample_A_R1.fastq.gz multiplex_sample_A_R2.fastq.gz --outFileNamePrefix zhou_2019/STAR_ALIGN_multiplex_sample_A.STARSOLO --outSAMtype BAM SortedByCoordinate --outSAMattributes NH HI nM AS CR UR CB UB GX GN sS sQ sM --soloCBstart 1 --soloCBlen 8 --soloUMIstart 9 --soloUMIlen 7 --soloCellFilter None --soloBarcodeReadLength 0’.
For female embryos, single cells were grouped by cell type and developmental day, according to the published sample annotation20,25 refined by clustering on the UMAP (uniform manifold approximation and projection) of batch-corrected data (‘3_buildModel.R’ in https://gitlab.univ-nantes.fr/E137833T/Castel_et_al_2020/-/blob/master/Code/). The sum of raw counts and counts per million (CPM) normalization were computed for each group. The median gene expression of the X chromosome and autosomes was calculated after removing nondetected genes.
SNP callingWe used a whole-genome sequencing dataset of H9 hES cells to identify informative genomic SNPs along the X chromosome that can be used for allelic expression analysis (GSM1227088). Reads were aligned using Bowtie 2 and PCR duplicates were filtered out using MarkDuplicates from Picard (version 2.22.0) as previously described. For proper tag formatting of BAM files, AddorReplaceReadGroups from Picard was used with the following options: ‘SO = coordinate, RGID = id, RGLB = library, RGPL = platform, RGPU = machine, RGSM = sample’. BAM files were further processed using GATK (version 4.1.2.0)59 for SNP identification. BaseRecalibrator and ApplyBQSR were used to generate the recalibrated BAM file using high-confidence SNPs referenced in Mills_and_1000G_gold_standard.indels.hg38.vcf.gz (annotation of indels) and dbsnp_146.hg38.vcf.gz (annotation of SNPs). The Variant Call Format (VCF) file H9_WGS_hg38_filtered.vcf was produced with HaplotypeCaller (options: ‘--dont-use-soft-clipped-bases and -stand-call-conf 10.0’) and filtered using VariantFiltration (options: ‘-window 50 -cluster 3 --filter-name FS -filter FS > 30.0 --filter-name QD -filter QD < 2.0’). The SNP position file info_snp_heterozygous_h9_wgs_chrX.txt was produced from VCF in R (version 4.1.1), using minimum thresholds of 50 for quality score, 10 for read coverage and 0.25 for allelic ratio to define heterozygous positions. We then used mpileup from SAMtools (version 1.15.1) to produce pileup files from RNA-seq and fastGRO-seq BAM files with the following options: ‘--output-MQ --no-output-ins --no-output-ins --no-output-del --no-output-del --no-output-ends’. The putative Xa haplotype was generated in R (version 4.1.1) from monoallelically expressed SNPs from the X chromosome in H9 primed hES cells. This was used to determine the pXi:Xa allelic ratio across all samples. We considered a transcript as biallelic when at least 25% of reads originated from the second allele.
XIST-sensitive and XIST-resistant gene callingWe applied stringent criteria for defining genes sensitive or resistant to XIST in naive cells. First, we determined the pXi:Xa allelic ratio for all detected SNPs, as described above, in XIST KO and CRISPRi datasets. We then computed the FC of the pXi:Xa allelic ratio between the XIST KO and WT and the XIST Dox-treated and UT CRISPRi datasets. An X-linked gene was defined as sensitive to XIST when the FC was greater than 1.2 in both datasets.
CUT&RUN data analysisBigWig track files were generated with bamCoverage from deepTools (version 3.5.0) using the following parameters: ‘--normalizeUsing BPM --binSize 20 --smoothLength 40’. Coverage score matrices were generated with computeMatrix from deepTools (version 3.5.0) using either reference-point (‘--referencePoint TSS -R Homo_sapiens.GRCh38.90.gtf --beforeRegionStartLength 1000 --afterRegionStartLength 1000 --binSize 20 --missingDataAsZero’) or scale-regions (‘-R Homo_sapiens.GRCh38.90.gtf --regionBodyLength 20000 --startLabel TSS --endLabel TES --binSize 20 --missingDataAsZero’). For histone modification enrichment, raw counts were generated using multiBigwigSummary from deepTools with ‘--binSize 10000’ and the mean counts per chromosome were calculated. The percentage occupancy was determined by calculating for each chromosome the cumulative coverage over the chromosome size. Regions with a minimum threshold of 1.2 (FC over IgG) were considered covered.
ATAC-seq data analysisATAC-seq raw data were analyzed using the atacseq pipeline (version 1.2.1) (https://github.com/nf-core/atacseq/) from nf-core60 using the default parameters. Briefly, reads were aligned to the human genome (hg38) using BWA (version 0.7.17). Duplicates were marked by Picard and reads mapping to mitochondrial DNA and blacklisted regions were removed. BigWig files were generated using deepTools as described above.
MeD-seq data analysisData processing was carried out using specifically created scripts in Python. Raw FASTQ files were subjected to Illumina adaptor trimming and reads were filtered on the basis of LpnPI restriction site occurrence 13–17 bp from either the 5′ or 3′ end of the read. Reads that passed the filter were mapped to hg38 using Bowtie 2. Genome-wide individual LpnPI site scores were used to generate read count scores for the following annotated regions: TSSs (1 kb before and 1 kb after), CpG islands and gene bodies (1 kb after TSS until TES (transcription end site)) and normalized (reads per million mapped reads (RPM)) using the total number of CpG reads after the filter. Gene and CpG island annotations were downloaded from ENSEMBL (www.ensembl.org).
RAPseq data analysisBigWig tracks were generated as previously described. RAPseq naive versus primed log2 enrichment was calculated using bigwigCompare from deepTools with the default parameters. The percentage occupancy was determined by calculating for each chromosome the cumulative coverage over the chromosome size. Regions with a minimum threshold of 10 (FC over input) were considered covered. RAPseq coverage scores were calculated with multiBigwigSummary, using the bins option for gene density enrichment and BED-file option for TEs (annotation from RepeatMasker).
Statistics and reproducibilityStatistical analyses were performed in R (version 4.1.1). The statistical analysis method for each experiment is specified in the corresponding figure legend. P values < 0.05 were considered statistically significant. Unless otherwise mentioned, most of the data shown are either representative of three or more independent experiments or combined from three or more biologically independent samples and analyzed as the mean ± s.d.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)