
記住我

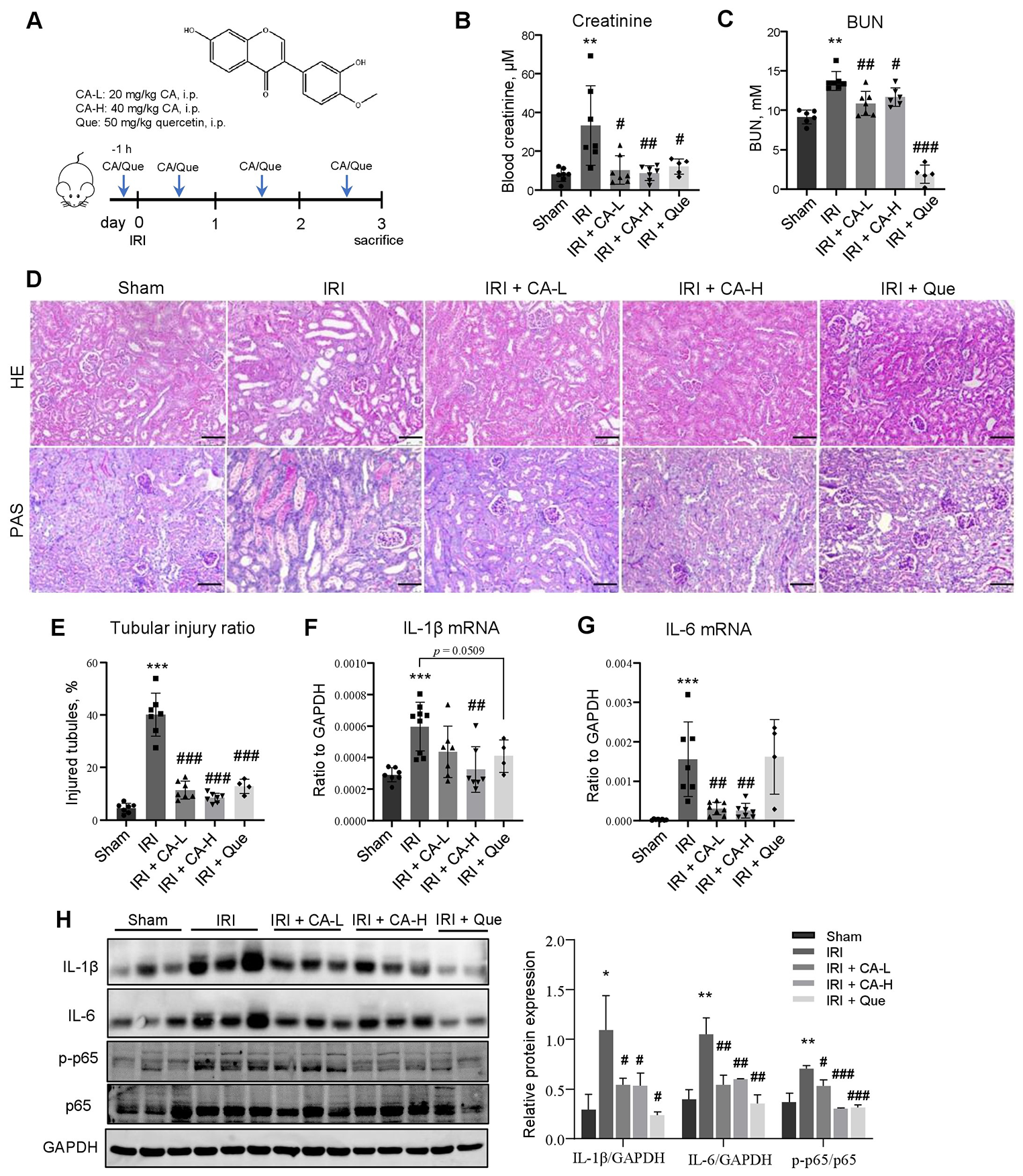
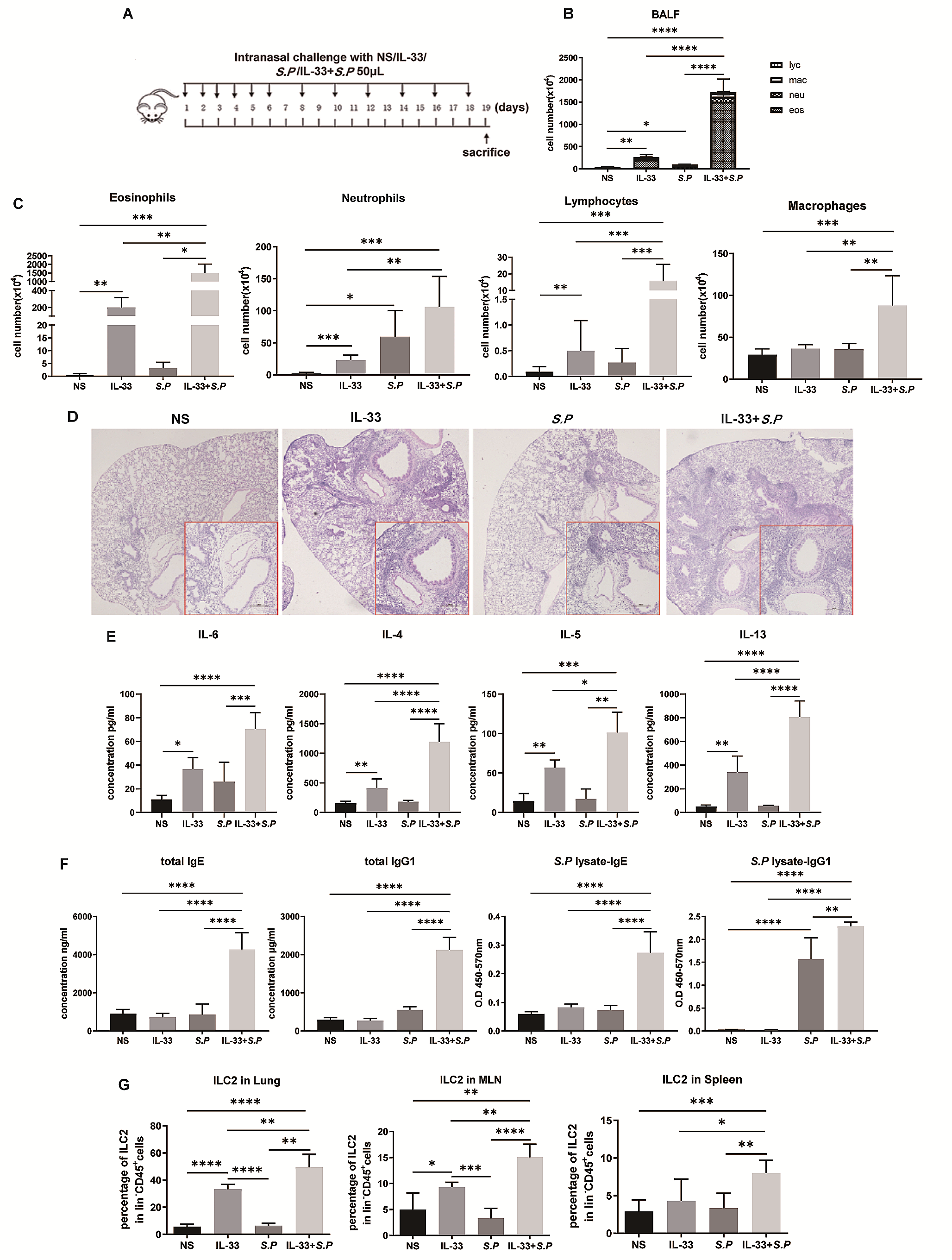
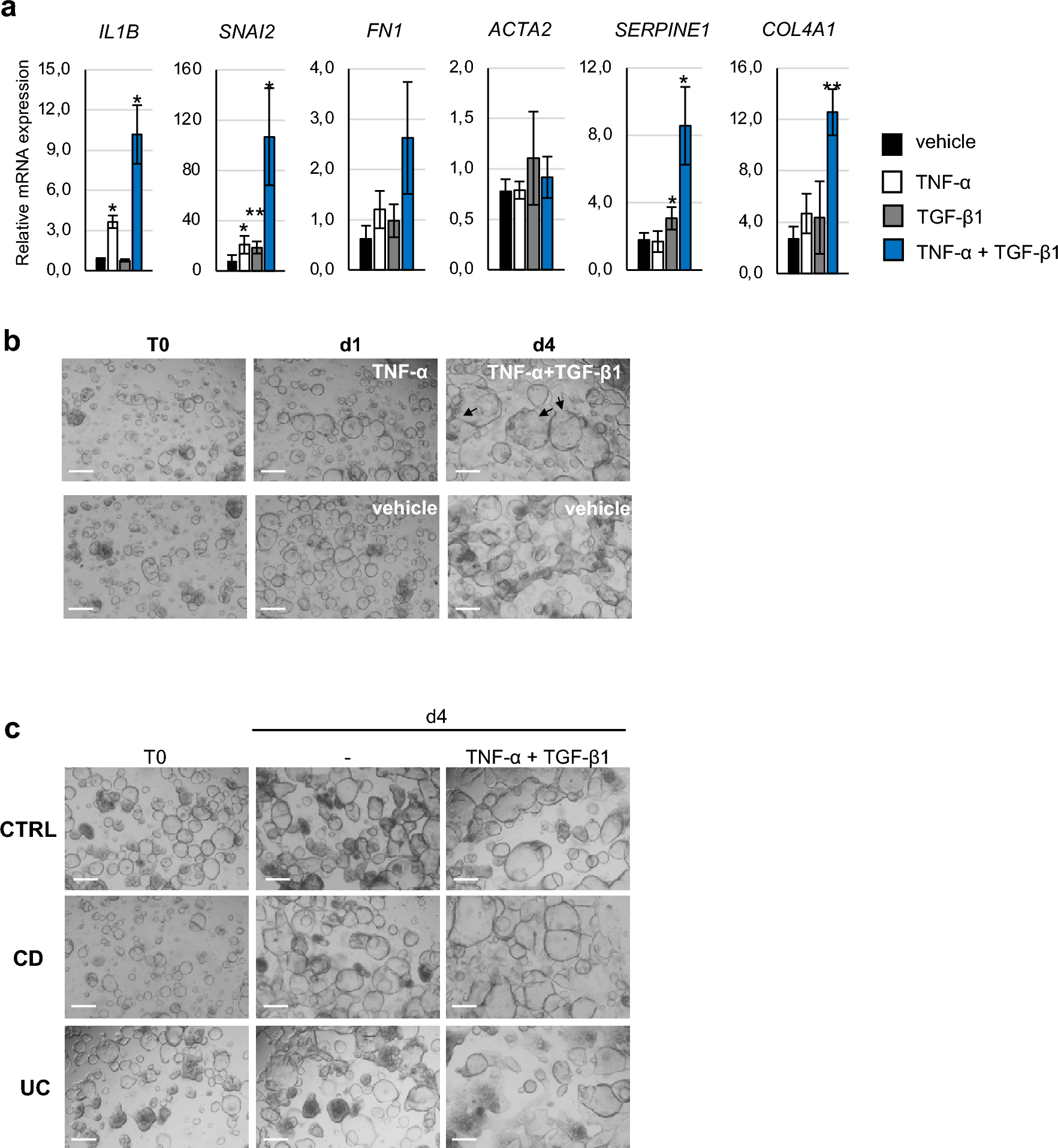
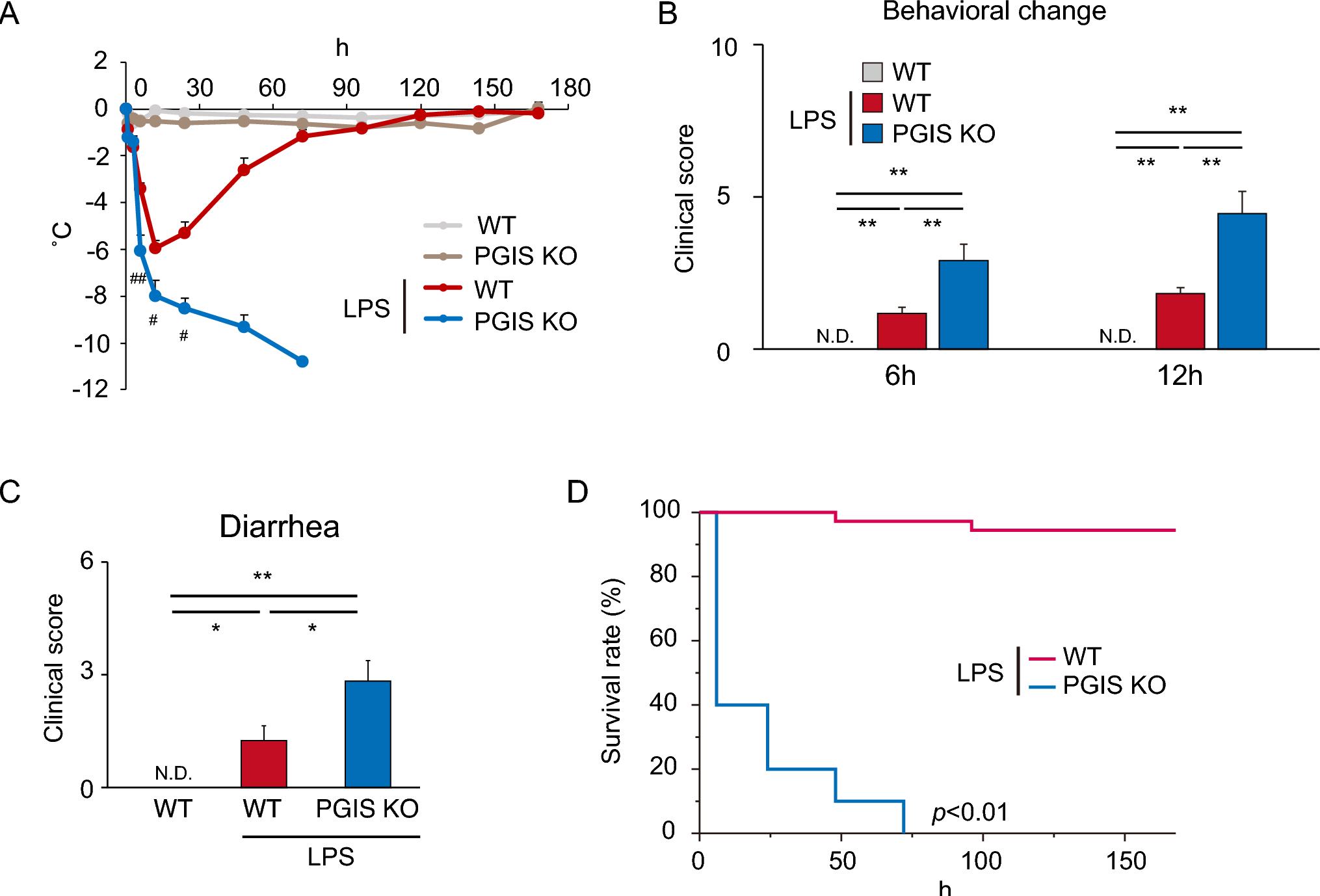
The immortalized human bronchial epithelial cell line 16HBE (Cat. BFN608008569) was purchased from Qingqi Biotechnology (Shanghai, China). LPS from Pseudomonas aeruginosa 10 (Cat. L8643) was purchased from Sigma‒Aldrich (Darmstadt, Germany). Exendin-4 (Cat. 141758-74-9) was purchased from MedChemExpress (New Jersey, USA). A Cell Counting Kit (CCK)‑8 (Cat. BS350B) was obtained from Biosharp (Anhui, China). Human TNF-α (Cat. ml077385), IL-1β (Cat. ml058059), and IL-6 (Cat. ml027379) and rat TNF-α (Cat. ml002859), IL-1β (Cat. ml058059), and IL-6 (Cat. ml102828) enzyme‑linked immunosorbent assay (ELISA) kits were obtained from Mlbio (Shanghai, China). An NF-κB activation-nuclear translocation assay kit (Cat. SN368) was purchased from Beyotime Biotechnology (Shanghai, China). The MiniBEST Universal RNA extraction kit (Cat. 9767), PrimeScript RT Reagent Kit with gDNA Eraser (Cat. RR047A), and TB Green Premix Ex Taq II (Cat. RR820A) were obtained from TaKaRa Bio (Beijing, China). Rabbit anti-T1R2 polyclonal (Cat. bs-9599R) and rabbit anti-T1R3 polyclonal antibodies (Cat. bs-23618R) for immunofluorescence and rabbit anti-β-actin polyclonal antibodies (Cat. bs-0061R) were purchased from Bioss (Beijing, China). Rabbit anti-T1R2 polyclonal (Cat. orb336464) and rabbit anti-T1R3 polyclonal (Cat. orb541918) antibodies for western blotting were obtained from Biorbyt (Cambridge, UK). Rabbit anti-NOD1 polyclonal antibody (Cat. P42567-1) was purchased from Abmart (Shanghai, China). Rabbit anti-NF-κB p65 polyclonal antibody (Cat. 80979-1-RR) was purchased from Proteintech (Wuhan, China). HRP-conjugated goat anti-rabbit IgG (Cat. GB23303) and CY3-conjugated goat anti-rabbit IgG (Cat. GB21303) were obtained from Servicebio (Wuhan, China). Alexa Fluor 488-conjugated goat anti-rabbit IgG (Cat. ab150077) was obtained from Abcam (Cambridge, UK).
Cell culture and groupingAll 16HBE cells were cultured in minimum essential medium (MEM, Cat. PM150410, Procell, Wuhan, China) supplemented with 5.6 mM glucose and 10% fetal bovine serum (Cat. 164,210, Procell, Wuhan, China) in a humidified incubator at 37 ℃ and 5% CO2 for 12 h, and experiments were subsequently conducted upon complete cellular adhesion.
First, for determination of the appropriate concentration of glucose for T1R2/T1R3, the cells were randomly grouped as follows (Fig. 1A): (1) the normal control (NC) group (with 5.6 mM glucose), (2) the 10 mM glucose group, (3) the 20 mM glucose group, and (4) the 30 mM glucose group. The glucose concentrations reported for all groups were the final concentrations including the known amount in the media used. All cells were cultured in an incubator at 37 ℃ for 24 h before the subsequent experiments.
Afterward, to investigate the effects and mechanisms of high glucose on LPS-induced airway epithelial inflammation, the cells were randomized into the following groups (Fig. 1B): (1) the normal control (NC) group (with 5.6 mM glucose for 24 h); (2) the NC + LPS group (after 12 h of preintervention with 5.6 mM glucose, 40 μg/ml LPS was added for an additional 12 h); (3) the 20 mM + LPS group (after 12 h of preintervention with 20 mM glucose, 40 μg/ml LPS was added for an additional 12 h); and (4) the 20 mM + LPS + Ex-4 group (after 10 h of preintervention with 20 mM glucose, 200 nM exendin-4, as previously described [29], was added for 2 h prior to 12 h of LPS exposure).
Fig. 1
Flow charts showing the design of the present cellular experiments. (A) Effect of different concentrations of glucose on airway epithelial 16HBE cells. (B) The effects of high glucose, lipopolysaccharide (LPS), and exendin-4 (Ex-4) on 16HBE cells. MEM: minimum essential medium; NC: normal control; mM: mmol/l
Cell viability assayThe ideal concentration of LPS isolated from PA was determined by a CCK-8 assay according to the manufacturer’s protocols. Briefly, 200 μl of 16HBE cell suspension at a density of 1 × 104 cells/ml was seeded into each well of a 96-well plate (Servicebio, Wuhan, China). Then, the cells were exposed to LPS at various concentrations (2.5, 5, 10, 20, 40, 80, 160, and 320 μg/ml) and maintained in a culture environment at 37 ℃ with 5% CO2. Each group had 5 replicate wells. After the intervention for 12 h, 10 μl of CCK-8 reagent was added to each well, and the cells were incubated at 37 °C for 1 h. Finally, the optical density (OD) of each well was measured at a wavelength of 450 nm using a microplate reader (Synergy HTX, BioTek, USA). Cell viability was assessed by the OD value.
Cellular immunofluorescenceEach group of cells was washed three times with phosphate-buffered saline (PBS) and then fixed at room temperature with 4% paraformaldehyde for 15 min. The cells were not permeabilized to reduce accidental staining of the cell nucleus. Afterward, the cells were blocked at room temperature with goat serum for 1 h. After the goat serum was removed, the cells were incubated with primary antibodies against T1R2 (dilution, 1:200), T1R3 (dilution, 1:200) or NOD1 (dilution, 1:200) at room temperature for 1 h. After being washed three times with PBS for 5 min each time, the cells were incubated with an Alexa Fluor 488-conjugated secondary antibody (dilution, 1:400) at room temperature for 1 h and with DAPI (Cat. C1006, Beyotime Biotechnology, Shanghai, China) for 5 min. After stained cells were imaged via confocal laser scanning microscopy (FV3000, Olympus, Japan), three images were selected for each group with different fields of view. The mean gray value was then analyzed using ImageJ software (version 1.8.0, National Institutes of Health, USA).
Cellular NF-κB activation-nuclear translocation assayAn NF-κB activation-nuclear translocation assay kit was used to assess NF-κB activation by detecting the translocation of the major subunit p65 into the nucleus through immunofluorescence staining. Notably, all reagents and antibodies used later came with the kit and did not require additional dilution. According to the assay kit protocol, after one wash with PBS, the cells were fixed with the fixative for 15 min and subsequently blocked with blocking solution for 1 h. Then, the cells were incubated with an NF-κB p65 primary antibody overnight at 4 ℃. After three 5‑min washes with the washing solution, the cells were incubated with secondary antibodies labeled with Cy3 at room temperature for 1 h. Finally, DAPI was used to stain the nuclei at room temperature for 5 min. After the stained cells were imaged using confocal laser scanning microscopy (FV3000, Olympus, Japan), three images were selected for each group with different fields of view. The mean gray value was then analyzed using ImageJ software (version 1.8.0, National Institutes of Health, USA).
Bacterial strains and culture conditionsThe Pseudomonas aeruginosa (PA) strain ATCC27853 was obtained as a generous gift from the Department of Laboratory Medicine of the First Affiliated Hospital of Hainan Medical University. As previously described [30], the bacteria were cultivated in Luria broth at 37 ℃ for 6 h until reaching the midlogarithmic phase. The bacteria were then extracted by centrifugation at 1500 × g for 15 min, washed twice in 0.9% NaCl without pyrogen, and resuspended in 10 ml of 0.9% NaCl. A total of 106 colony forming units (CFU) per ml of PA suspension was used for intratracheal instillation.
Animal model preparation and groupingSprague‒Dawley (SD) adult male rats (230–270 g) were purchased from Hunan Slack Jingda Company (Hunan, China). The rats were provided unlimited access to food and fresh water. The rats were acclimated to a room temperature of 22–25 ℃, and three rats were housed per cage. A light/dark cycle of approximately 12 h was maintained. All in vivo procedures adhered to the National Institutes of Health (NIH) guidelines and received approval from the Ethical Committee of Hainan Medical University (Code: HYLL-2021-218). The 25 SD rats were randomly divided into five groups as follows: (1) the normal control (NC) group (with a normal diet); (2) the PA group, in which the rats were anesthetized with an intraperitoneal injection of ketamine (20 mg/kg) and subsequently given an intratracheal instillation of 50 μl of bacterial suspension, as previously described [31]; (3) the diabetes mellitus (DM) group, in which the rats were fed a high-fat diet composed of 45% fat, 35% carbohydrates and 20% protein for 6 weeks, followed by an intraperitoneal injection of streptozotocin (50 mg/kg), with high blood glucose levels exceeding 250 mg/dl as the criteria for diabetes diagnosis, as previously described [32]; (4) the DM + PA group, in which rat models were initially established for diabetes and subsequently for PA-related pneumonia; and (5) the DM + PA + Ex-4 group, in which after being induced with diabetes, rats were initially pretreated with 10 μg/kg EX-4 intraperitoneally twice a day for 7 days, as previously described [33], then infected by PA, and finally treated with the same dose of exendin-4 for another 7 days (Fig. 2).
Fig. 2
The diabetes mellitus (DM) model, Pseudomonas aeruginosa (PA) pulmonary infection model and PA-infected DM model were established, as was the exendin-4 (Ex-4) treatment process. NC: normal control; STZ: streptozotocin; CFU: colony forming units
Bronchoalveolar lavage fluid (BALF) and lung tissue preparationAs previously described [34], all rats were anesthetized through the intraperitoneal administration of ketamine (20 mg/kg), after which a thoracotomy was performed. After the left lung bronchioles were ligated with surgical sutures, 3 ml aliquots of normal saline were slowly infused into the lungs through tracheostomy and then withdrawn gently. This process of lavage was reiterated thrice utilizing the same syringe. The pooled BALF was centrifuged at 500×g at 4 °C for 5 min. The cell-free supernatants were stored in microcentrifuge tubes at -80 °C for subsequent ELISA analysis. Finally, the left lungs were inflated and fixed with 10% formalin overnight at room temperature for subsequent experiments, and the right lungs were preserved in liquid nitrogen.
Hematoxylin and eosin (HE) stainingThe left lung tissues of the rats were collected and fixed with 10% formalin. Subsequently, the tissues were dehydrated, embedded in paraffin, and sliced into thin 5 μm slices. These sections were then stained with hematoxylin and eosin. Finally, the pathological alterations in the tissues were evaluated using a section scanner (Pannoramic MIDI, 3DHISTECH, Hungary).
Immunofluorescence staining of lung tissue paraffin sectionsAs previously described [35], the lung tissue paraffin sections were deparaffinized and rehydrated, followed by antigen repair. Then, the slides were blocked with 5% donkey serum at room temperature for 30 min. Primary antibodies against T1R2 (dilution, 1:100), T1R3 (dilution, 1:100), NOD1 (dilution, 1:100) or NF-κB p65 (dilution, 1:250) were incubated with the slides at 4 ℃ overnight. Then, the slides were placed in PBS and shaken 3 times for 5 min each time. After that, the sections were incubated with secondary antibodies conjugated to Alexa Fluor 488 (dilution, 1:400) or CY3 (dilution, 1:300) at room temperature for 50 min in the dark. DAPI solution was added dropwise, and the cells were incubated at room temperature for 10 min in the dark. Finally, the slides were observed using a fluorescence microscope (Eclipse C1, Nikon, Japan).
ELISAs of cellular and BALF supernatantsThe levels of TNF-α, IL-1β and IL-6 in the extracted cellular and BALF supernatants were evaluated using the corresponding kits according to the instructions. The OD was assessed using a microplate reader (Synergy HTX, BioTek, USA) at a wavelength of 450 nm.
Reverse transcription‑quantitative PCR (RT‑qPCR) of cells and lung tissuesTotal RNA was extracted from cells and lung tissues using the TaKaRa MiniBEST Universal RNA Extraction Kit following the manufacturer’s protocols. After that, reverse transcription was performed using the TaKaRa PrimeScript RT Reagent with gDNA Eraser kit. Finally, the mRNA expression levels of TNF-α, IL-1β and IL-6 were measured using a TaKaRa TB Green Premix Ex Taq II kit. The primer sequences, which were designed using Primer Premier 5.0 software, are displayed in Table 1. The mRNA levels were quantified using the 2‑ΔΔCq method [36]. All groups had at least 3 repetitions.
Table 1 Sequences of primers used in the present studyWestern blot analysis of cells and lung tissuesTotal protein was extracted from the cells and lung tissues using RIPA buffer supplemented with PMSF. The quantification of total protein was conducted using a BCA assay, followed by the separation of proteins on a 5–10% SDS‒PAGE gel. A volume of 20 μL of protein was sampled from each sample. Equal quantities of protein from each sample were subsequently transferred onto PVDF membranes. The membranes were then blocked with PBS with 0.05% Tween-20 and 5% nonfat milk for 1 h. Thereafter, the membranes were incubated with primary antibodies against T1R2 (diluted 1:2000), T1R3 (diluted 1:2000), NOD1 (diluted 1:2000), NF-κB p65 (diluted 1:5000) or β-actin (diluted 1:5000) at 4 ℃ overnight. After three washing cycles, the membranes were incubated with an HRP-conjugated secondary antibody (diluted 1:5000) for 2 h at room temperature. The protein bands were visualized after an additional three washes using an enhanced chemiluminescence reagent. The protein expression levels were quantified using ImageJ software (version 1.8.0, National Institutes of Health, USA), with β-actin serving as the loading control.
Statistical analysisAll the data are presented as the mean ± standard deviation, and at least 3 repetitions were performed. The various groups were statistically analyzed through one-way analysis of variance (ANOVA), which was subsequently followed by Tukey’s post hoc test. All the data were analyzed using GraphPad Prism 9 (GraphPad Software, USA). p < 0.05 was considered to indicate a statistically significant difference.
留言 (0)