
記住我



Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of disorders characterized by defective secretion of bile acids or transport defects resulting in progressive cholestasis. These disorders usually present during infancy or childhood and are associated with progressive liver disease.1 PFIC is estimated to affect 1 in 50,000–100,000 births. Based on clinical presentation, laboratory findings, liver histology, and genetic defect, these are broadly divided into 3 types—PFIC1, PFIC2, and PFIC3. The defect is in ATP8B1 gene encoding the FIC1 protein, ABCB 11 gene encoding the Bile Salt Export Pump (BSEP) protein, and ABCB4 gene encoding the MDR3 protein in PFIC1, 2, and 3, respectively. The basic defect is impaired bile salt secretion in PFIC1/2, whereas in PFIC3, it is reduced biliary phospholipid secretion. The main clinical presentation is in the form of cholestatic jaundice and pruritus. Serum gamma glutamyl transpeptidase (GGT) is normal in patients with PFIC1/2 while it is raised in patients with PFIC3.1,2
PFIC2 represents half of PFIC cases and presents with hepatosplenomegaly, jaundice, pruritus, fat-soluble vitamin deficiencies, and growth failure. Laboratory findings in PFIC2 include low/normal GGT levels, elevated bilirubin, elevated transaminases, and higher alpha-fetoprotein levels.3 In this report, we present a case of PFIC2 presenting with severe coagulopathy, bruising, subcutaneous hematomas, and acute-onset anemia.
CASE REPORTOur patient is a previously healthy 8-month-old girl who presented to the hospital with a 3-day history of spontaneous bruising over extremities, lower back, abdomen, and neck. The mother also noted pallor and decreased activity and areas of soft-tissue swelling beneath the bruises on the lower back and abdomen. She denied pale stools, but reported 2 episodes of melena, a day before admission. The child had no history of easy bruising or nose bleeding. She was on a regular infant formula and baby foods with no restrictions. She was otherwise healthy, growing well with normal developmental milestones. She was not on any home medications or multivitamin supplementation.
Pregnancy was uncomplicated, and she was born full term at 39 weeks gestation and received vitamin K injection at birth. Physical examination was notable for diffuse areas of excoriation secondary to pruritus, mild scleral icterus, hepatomegaly, but no splenomegaly. There were multiple bruises of varying sizes and discoloration noted on the neck crease, right upper chest, left axilla, flanks, lower back, and lower abdomen and extremities (Figures 1–4). Owing to concerns for possible underlying liver disease, further history was elucidated. The mother reported a history of mild pruritus, but no history of neonatal jaundice. Newborn screening result was normal including negative screening for cystic fibrosis. Extended family history revealed no concerns for bleeding disorders or genetic conditions. Social history was concerning for maternal drug use and prior Department of Social Services (DSS)/Child Protective Services involvement for possible child neglect.
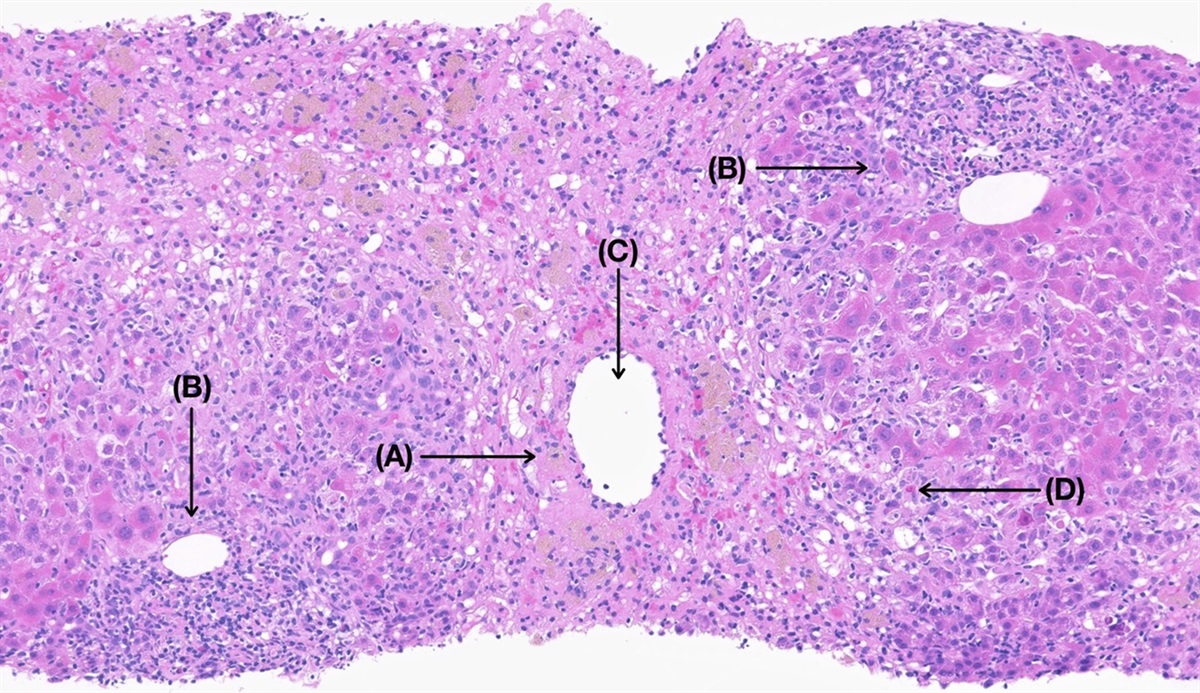
 Figure 1.:
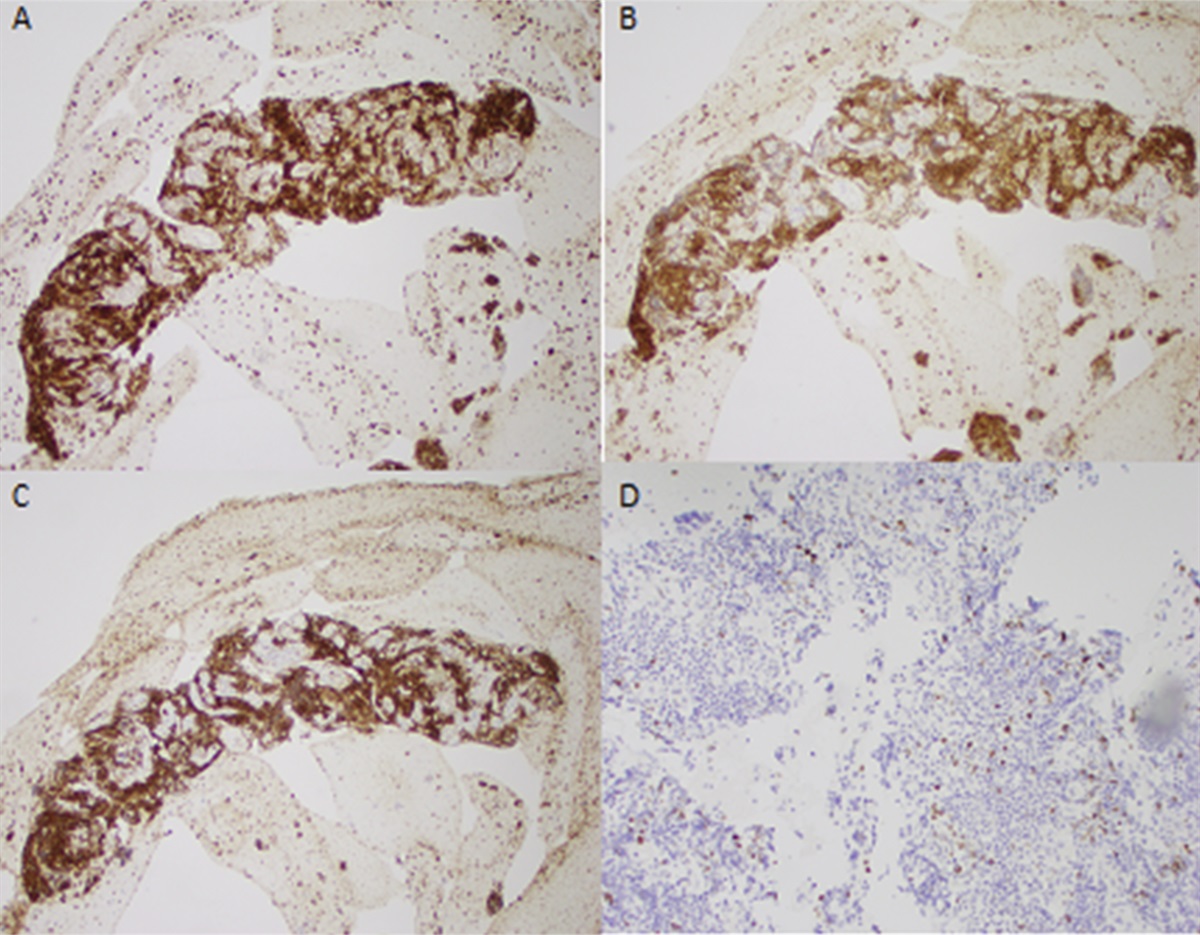
Figure 1.: (A) H&E stain: low-power giant cells (black arrows) with cholestasis (blue arrows). (B) Trichrome stain: low-power perisinusoidal fibrosis (blue arrow) with expansion of portal tracts (yellow arrow).
 Figure 2.:
Figure 2.: Multiple bruises on the neck and upper chest.
 Figure 3.:
Figure 3.: Multiple bruises on the axilla and abdomen.
 Figure 4.:
Figure 4.: Multiple bruises on the lower back.
Blood work revealed severe anemia (hemoglobin 3.9 gm/dL, White Blood Cell (WBC) count at 20 k, and platelet count 354 k), coagulopathy (International Normalised Ratio (INR) >14, Partial Thromboplastin Time (PTT) >180), and elevated total bilirubin (3.5 mg/dL, direct bilirubin at 2.1 mg/dL), alanine aminotransferase 199 U/L, and aspartate aminotransferase 293 U/L. Alkaline phosphatase was 290 U/L, and Gamma Glutamyl Transferase (GGT) was normal at 13 U/L. Fresh frozen plasma was administered in the emergency department, followed by 5 mg of vitamin K injection, resulting in the correction of INR. Protein induced by vitamin K absence-II (PIVKA-II) testing to confirm Vitamin K deficiency could not be obtained at our institution prior to Vitamin K administration. Abdominal ultrasound revealed a normal liver and spleen.
The child was admitted to the hospital for further evaluation. Multiple teams were involved in care, including gastroenterology, toxicology, genetics, hematology/oncology, and child maltreatment teams.
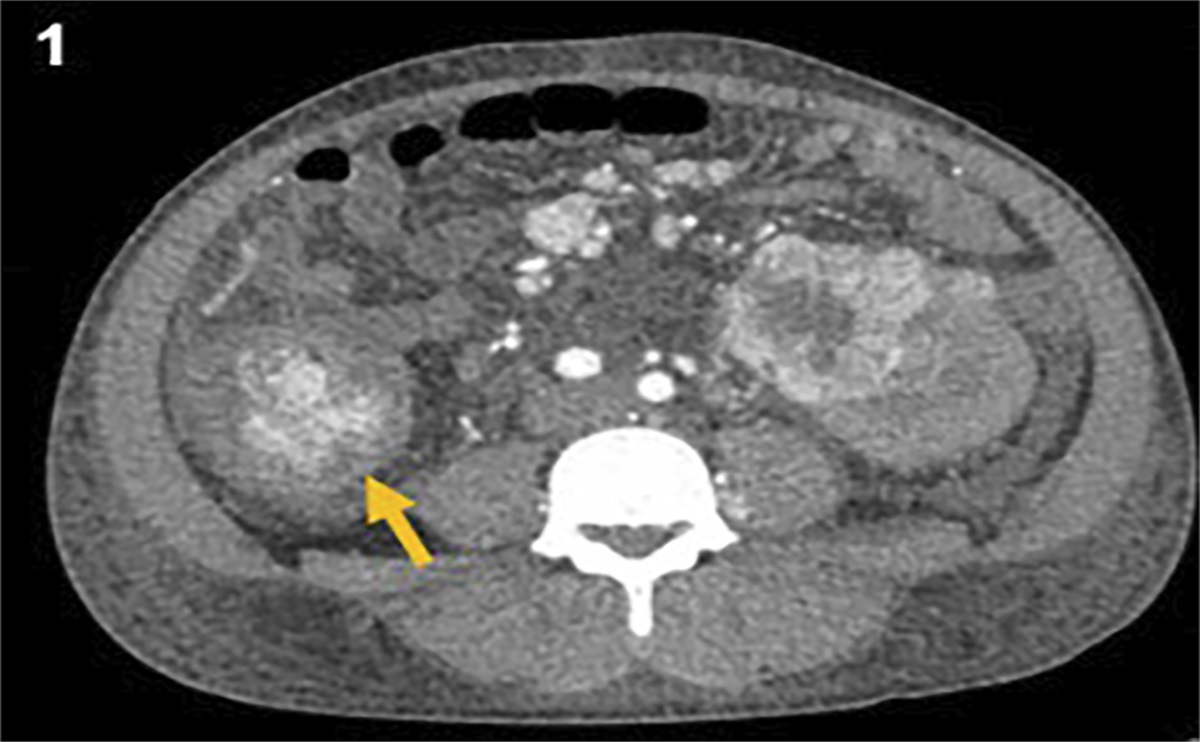
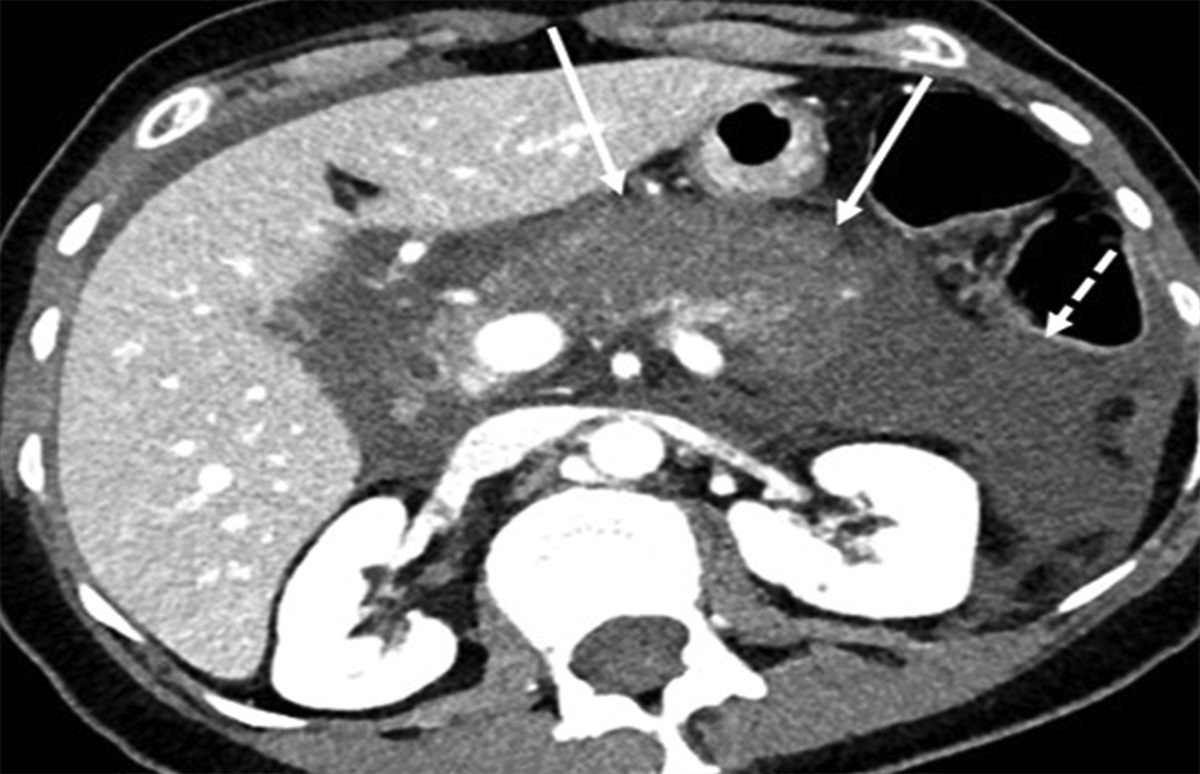
With multiple large bruises and hematomas on the abdomen and back, child abuse was in the differential. Skeletal survey and Computed Tomography Scan of chest and abdomen obtained due to concerns for non-accidental trauma were negative. The DSS was reconsulted, and the infant was placed in foster care pending further investigation.
Given the vitamin K-responsive coagulopathy, concern was raised for warfarin or rodenticide accidental ingestion, and the toxicology team was consulted but was subsequently ruled out. There were no family members on warfarin therapy, and the family did not have any rodenticides at home.
Acute leukemia was in the differential in the child with acute-onset anemia, easy bruising and severe coagulopathy. Peripheral blood smear was normal. Once the child was stabilized and coagulopathy corrected, bone marrow aspirate was performed that showed no abnormalities.
Workup for likely cholestatic liver disorders was initiated with history of cholestatic jaundice, pruritus, and vitamin K malabsorption, leading to coagulopathy. The alpha-1 antitrypsin phenotype was normal. Alpha-fetoprotein was elevated at 412 ng/mL (normal <9.0) as were triglycerides at 235 mg/dL (normal <70). Serum bile acids were noted to be markedly elevated at 482 μmol/L (normal <9.2). Vitamin D, vitamin E, and vitamin A levels were low. Diagnostic liver biopsy revealed giant cell hepatitis with canalicular cholestasis and stage 2–3 periportal and sinusoidal fibrosis with focal bridging. Genetic testing using rapid whole-exome sequencing found compound heterozygous likely pathogenic variants in ABCB11 (c.2782_2783insGAGAT c.2012-8T>G) consistent with a diagnosis of PFIC2. The DSS case was closed, and the child was returned to the family.
The child was started on Fat soluble vitamins ADEK and ursodeoxycholic acid, and with refractory pruritus at 10 months of age, ileal bile acid transporter inhibitor (IBAT) therapy was initiated. Her pruritus improved minimally after initiation of IBAT therapy. At 18 months of age, her pruritus persists and liver parameters remain abnormal with a total bilirubin of 3.4 mg/dL, direct bilirubin 2.2 mg/dL, alanine aminotransferase 272 U/L, aspartate aminotransferase 229 U/L. Alkaline phosphatase is 274 U/L. Total bile acid levels remain high at 347 μmol/L. Prothrombin Time (PT) and INR are normal at 13.5 and 1.0, respectively. Vitamin A level is normal at 25 μg/dL, vitamin D level low at 16 ng/mL, and vitamin E level low at <0.4 mg/L despite supplementation. Her alpha-fetoprotein levels have improved but high (88 ng/mL). She will be monitored long term for progression of liver disease to cirrhosis and possible hepatocellular carcinoma.
DISCUSSIONPFIC is a heterogeneous group of rare autosomal recessive liver disorders of childhood characterized by mutations in genes encoding proteins involved in the hepatocellular transport system. Multiple symptoms are reported at presentation, including jaundice, hepatomegaly, itching/pruritus, pale stools, splenomegaly, diarrhea, discolored stools, failure to thrive, vitamin E deficiency, vitamin D deficiency, and pancreatitis specific to PFIC1.4–8 The median age of onset for PFIC1 and PFIC2 is 3 months of life, with a tendency for earlier appearance in patients with PFIC2.8 Some patients with PFIC2 also present with early signs of vitamin D deficiencies (rickets, 3%–22%), vitamin K deficiencies (bleeding, 8%), or cholelithiasis (28%).4,5 There are cases reported with PFIC2 presenting with intracranial bleeding secondary to vitamin K deficiency mimicking abusive head injury.9
PFIC2 is caused by biallelic pathogenic mutations in ABCB11 gene, which encodes the bile salt export pump (BSEP) protein. The BSEP protein is involved in actively transporting bile salts out of hepatocytes into biliary canaliculi. Mutations in this gene interrupt the process of pumping bile out of hepatocytes, leading to increased intracellular bile salt concentrations, which consequently damage the hepatocytes. Liver histology reveals canalicular cholestasis, the absence of true ductular proliferation with only periportal biliary metaplasia of hepatocytes, pronounced lobular and portal fibrosis and inflammation, hepatocellular necrosis, and giant cell transformation. BSEP immunostaining is helpful for diagnosis. Genotyping confirms the diagnosis.10 There is up to a 15% chance of cirrhosis developing into either hepatocellular carcinoma or cholangiocarcinoma in patients with PFIC2.
There are pharmacologic treatment options for patients with PFIC that relieve symptoms or prevent disease progression. Treatments include ursodeoxycholic acid, IBAT, and agents for the symptomatic relief of pruritus such as antihistamines and rifampin (rifampicin).1,11 Not all patients respond to medical treatment and invasive surgery, such as ileal bypass, partial external biliary diversion, or partial internal biliary diversion may be needed to lower circulating bile acid concentrations.1,11,12 Ultimately, liver failure and intractable pruritus may indicate a need for liver transplantation (LT), most often in patients with PFIC2.8 The indications for LT in PFIC include severe pruritus, significant growth retardation, liver cirrhosis, and liver failure.1,13 The role of LT in PFIC2 is well documented with favorable outcomes, but the recurrence of disease secondary to anti-BSEP antibodies to donor liver is known.14
This case reminds us that it is important to rule-out medical diseases that can mimic child abuse, as misdiagnoses can further harm children and families. Children presenting with cholestasis and significant bruising should have congenital cholestatic disorders, such as PFIC, included in the differential diagnosis.
DISCLOSURESAuthor contributions: S. Reddy wrote the manuscript, reviewed the literature, and edited the manuscript. VV Gopalareddy, K. Dempsey, E. Ferren, and N. Fleishman revised the manuscript for intellectual content; M. Kamionek provided the images. VV Gopalareddy provided the intellectual content, edited and approved the final manuscript, and submitted the manuscript. V. Gopalareddy is the article guarantor.
Financial disclosure: None to report.
Previous presentation: NASPGHAN Annual Meeting; October 4–7, 2023; San Diego, California.
Informed consent was obtained for this case report.
REFERENCES 1. Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4(1):25–36. 2. Baker A, Kerkar N, Todorova L, Kamath BM, Houwen RHJ. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2019;43(1):20–36. 3. Agarwal S, Lal BB, Rawat D, Rastogi A, Bharathy KGS, Alam S. Progressive familial intrahepatic cholestasis (PFIC) in Indian children: Clinical spectrum and outcome. J Clin Exp Hepatol. 2016;6:203–8. 4. Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51(5):1645–55. 5. Al Mehaidib A, Al Shahrani A. Progressive familial intrahepatic cholestasis in Arabs. J Hepatol. 2013;58:S555–6. 6. Englert C, Grabhorn E, Richter A, Rogiers X, Burdelski M, Ganschow R. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation. 2007;84(10):1361–3. 7. Lee WS, Chai PF, Looi LM. Progressive familial intrahepatic cholestasis in Malaysian patients: A report of five cases. Med J Malaysia. 2009;64(3):216–9. 8. Wanty C, Joomye R, Van Hoorebeek N, et al. Fifteen years single center experience in the management of progressive familial intrahepatic cholestasis of infancy. Acta Gastroenterol Belg. 2004;67(4):313–9. 9. Haney S, Harper J, Truemper E. Progressive familial intrahepatic cholestasis presenting with an intracranial bleed and mimicking abusive head trauma. WMJ. 2019;118(1):47–8. 10. Vitale G, Gitto S, Raimondi F, et al. Cryptogenic cholestasis in young and adults: ATP8B1, ABCB11, ABCB4, and TJP2 gene variants analysis by high-throughput sequencing. J Gastroenterol. 2018;53(8):945–58. 11. Van der Woerd WL, Houwen RH, van de Graaf SF. Current and future therapies for inherited cholestatic liver diseases. World J Gastroenterol. 2017;23(5):763–75. 12. Bustorff-Silva J, Sbraggia Neto L, Olímpio H, et al. Partial internal biliary diversion through a cholecystojejunocolonic anastomosis: A novel surgical approach for patients with progressive familial intrahepatic cholestasis: A preliminary report. J Pediatr Surg. 2007;42(8):1337–40. 13. Zhu Z, Wei L. Liver transplantation for progressive familial intrahepatic cholestasis. 2018;23:666–73. 14. Mehl A, Bohorquez H, Serrano M-S, Galliano G, Reichman TW. Liver transplantation and the management of progressive familial intrahepatic cholestasis in children. World J Transpl. 2016;6(2):278–90.
留言 (0)