Proliferation, osteogenic and adipogenic induction of human MSCs
hBMMSCs and hASCs were purchased from Science Cell Research Laboratories (San Diego, CA). hBMMSCs were cultured in proliferation medium (PM) containing 10% FBS, α-MEM, and 1% streptomycin-penicillin. hASCs were cultured in PM containing 10% FBS, DMEM, and 1% streptomycin-penicillin. Osteogenic medium (OM) was standard PM with 0.2 mmol/L ascorbic acid, 10 mmol/L β-glycerophosphate, and 10 nmol/L dexamethasone. Adipogenic medium (AM) was standard PM with 200 μmol/L indomethacin, 100 nM dexamethasone, 50 nmol/L insulin and 500 μmol/L 3-isobutyl-1-methylxanthin.
Induction of apoptosis, isolation, and identification of apoVs
Culture medium was substituted for α-MEM/DMEM with 500 nmol/L STS (Enzo Life Sciences) to induce apoptosis of MSCs. MSCs were labeled using the TUNEL Apoptosis Detection Kit (Applygen, C000320) and rhodamine fluorescein (red) dUTP, and apoptosis was observed using a fluorescence microscopy (Olympus). After 12 h, apoVs were extracted by differential centrifugation (Fig. 1b)39. The Pierce BCA Assay Kit (Thermo Scientific) was used to quantify the apoVs concentration.
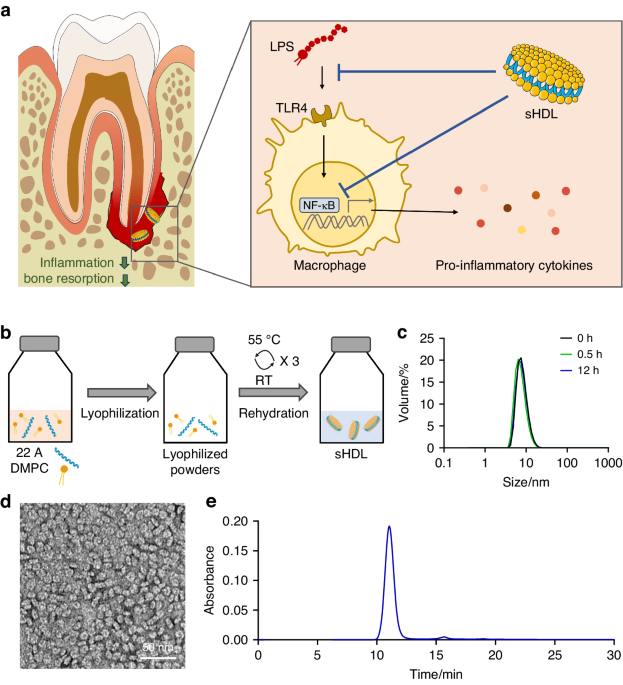
ApoVs were deposited onto a carbon-coated copper net, stained twice with 1% uranyl acetate, and their morphology was visualized using an HT7700 transmission electron microscopy (Hitachi). The size distributions were evaluated using the Nano Sight NS300 (Malvern) according to the manufacturer’s instructions. ApoV markers (CD9, CD81, CD63, Fas, calreticulin, CD44, and integrin α-5) were detected by Western blotting.
Assay of apoV uptake in vitro
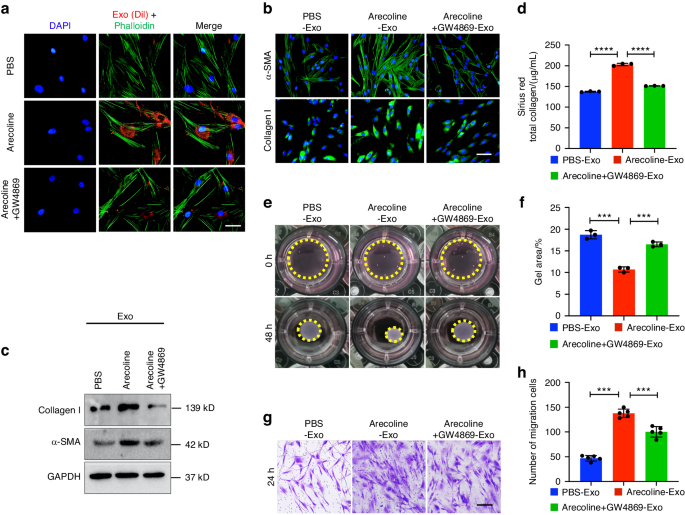
ApoVs were labeled with PKH-26 using the Red Fluorescent Cell Labeling Kit (Umibio). Next, they were washed and the supernatant was collected as the negative control (NC). Labeled apoVs and NC (250 ng/mL) were incubated with MSCs for 12 or 24 h, and 5 μg/mL phalloidin (Sigma-Aldrich) and 6-diamidine-2-phenylindole (DAPI) were added. Fluorescence imaging was performed using a confocal LSM 5 EXCITER microscope (Carl Zeiss).
Isolation of exosomes
Exosomes were removed from fetal bovine serum (FBS) by ultracentrifugation at 120 000 g for 18 h. Cells were cultured in exosome-free medium for 2 days, and the supernatant was centrifuged at 300 g for 10 min, 3 000 g for 10 min, and 20 000 g for 30 min. Leftover supernatant was passed through a 0.22 μm filter (Millipore) and ultracentrifuged at 120 000 g for 120 min to isolate exosomes.
Lentivirus infection
Recombined lentivirus overexpressing hsa-miR-4485-3p (mimics) and the NC (mi-NC) were purchased from GenePhama (Suzhou, China). The sequences were: mimics: 5’-TAACGGCCGCGGTACCCTAA-3’; mi-NC: 5’-TTCTCCGAACGTGTCACGT-3’. For transfection, cells at 30%–40% confluence were exposed to the viral supernatant. After 2 days, 1 mg/mL puromycin (Sigma-Aldrich) was added to select stably transfected cells.
Transient infection with miRNAs and plasmids
An RNA oligo with a 2’-ome modification to knockdown hsa-miR-4485-3p (inhibitor) and its NC (inhi-NC) were obtained from GenePhama (Suzhou, China). The sequences were: inhibitor: 5’-UUAGGGUACCGCGGCCGUUA-3’; inhi-NC: 5’-CAGUACUUUUGUGUAGUACAA-3’. The cells were transfected using Lipofectamine 3000 (Invitrogen).
Cell counting kit-8 assay
MSCs were cultured in PM or PM supplemented with 62.5, 125, 250, and 500 ng/mL MSC-apoVs. MSC viability of MSCs was evaluated with the Cell Counting Kit-8 (CCK8, Dojindo Laboratories, Kumamoto, Japan) at days 0, 1, 3, 5, and 7 in triplicate wells. The optical density at 450 nm were measured using the ELX808 Plate Reader (BioTek).
Alkaline phosphatase (ALP) and Alizarin red S (ARS) staining and quantification
MSCs were fixed in 95% ethanol. For ALP staining, the BCIP/NBT Staining Kit (CoWin Biotech) was used. ALP activity was measured based on the absorbance at 520 nm using the ALP Assay Kit (Nanjing Jiancheng Bioengineering Institute). For ARS staining, MSCs were incubated with 40 mmol/L filtered 2% Alizarin red buffer (Sigma-Aldrich), followed by 100 nmol/L cetylpyridine for >1 h to quantify calcium-bound ARS by measuring spectrophotometrically the absorbance at 562 nm.
Oil red O staining and quantification
MSCs were fixed in 10% neutral formalin and washed with 60% isopropanol. Next, Oil red O working solution (Sigma-Aldrich) was added and MSCs were observed using a microscope. Lipid droplets were dissolved in 100% isopropanol for quantitative evaluation, and the absorbance at 500 nm was measured.
RNA isolation and qRT-PCR
Total RNA was extracted from MSCs using TRIzol (Invitrogen, Carlsbad, CA). RNA was extracted from apoVs and exosomes using the miRNeasy Mini Kit (Qiagen). RNA purity and concentration were measured using a NanoDrop 8000 spectrophotometer (Pierce Thermo Scientific, Waltham, MA). The PrimeScript RT Reagent Kit (#RR037A; Takara, Tokyo, Japan) was used to produce cDNA. For miRNA evaluation, the miDETECT A TrackTM qRT-PCR Starter Kit (Ribo Bio) was used to produce cDNA. qRT-PCR was conducted using SYBR Green Master Mix (Yeasen Biotechnology, Shanghai, China) on the ABI Prism 7500 Real-Time PCR Detection System (Applied Biosystems, Foster City, CA). The sequences of the primers are listed in Table S4. Expression levels were normalized to those of GAPDH and U6.
Western blotting
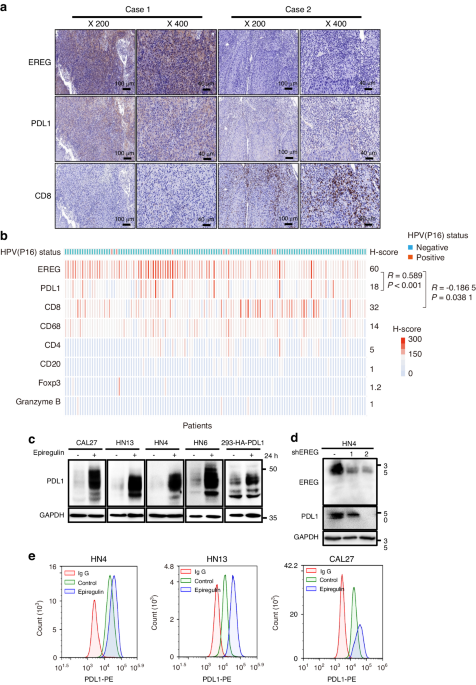
MSCs were lysed and the protein concentration was determined using the Pierce BCA Protein Assay Kit. Aliquots (40 μg) of protein solutions were resolved by 10% SDS-PAGE (Millipore, Billerica, MA) and transferred to PVDF membranes. The membranes were incubated with diluted anti-RUNX2, anti-PPARγ, anti-GAPDH, anti-β-catenin (ProteinTech), anti-CD9, anti-CD81, anti-Fas, anti-integrin alpha-5, anti-CD44, anti-p65 (Abcam), anti-calreticulin (Cell Signaling Technology), anti-AKT1 (Santa Cruz), anti-p-AKT1, anti-ERK and anti-p-ERK (Abclonal) primary antibodies, followed by the corresponding secondary antibodies (Cell Signaling Technology). Bands were detected using the ECL Kit (NCM bio, Suzhou, China). Images were analyzed using Image J software (National Institutes of Health, Bethesda, MD).
In vivo implantation of hMSCs
This experiment was approved by the Institutional Animal Care and Use Committee of the Peking University Health Science Center (approval number: LA2021006).
For heterotopic adipose tissue formation, MSCs were cultured for 7 days and seeded on collagen membrane scaffolds (Wuxi Biot Bioengineering Institute, Wuxi, China). For ectopic bone tissue formation, MSCs were combined with β-TCP (RB-SK-005 G). After anesthetization with pentobarbital, the hybrids were imbedded subcutaneously into the backs of nude mice. The implants were harvested after 6 weeks and fixed.
Mouse model of osteoporosis mouse model and tail-vein injection of apoVs
Eight-week-old female BALB/c mice underwent bilateral OVX to establish a mouse model of estrogen deficiency-induced bone loss. Other eight-week-old female BALB/c mice underwent a sham operation. Eight weeks after surgery, apoV solution (20 μg per 30 g body weight) and PBS (volume equal to that of apoV solution) were administered weekly to the mice via the tail vein. Twelve-month-old female mice also underwent tail-vein injection. The mice were humanely euthanized after 8 weeks and the femurs were dissected and fixed.
Analysis of apoV distribution in vivo
ApoVs were incubated with 3,3’-dioctadecyloxacarbocyanin perchlorate (DiR, Keygen Biotech, Jiangsu, China) solution and injected into mice via the tail vein. After 24 and 48 h, the livers, lungs, spleens, hearts, kidneys, spines, femurs, mandibular bones, and cranial bones were harvested and scanned using the IVIS Lumina Series III (PerkinElmer, MA).
Restoration of cranial defects in rat using apoV-PLGA/pDA scaffolds
To produce apoV-PLGA/pDA scaffolds, PLGA/pDA scaffolds were immersed in 1 μg/μL apoV solution for 12 h. The scaffolds were immersed in normal saline, which was replenished daily for assessment of apoVs release by BCA assay. Rats with 5 mm diameter skull defects were subjected to the indicated treatments. After 8 weeks, the cranial bones were harvested and fixed.
Histological staining and assessment
After fixation for at least 2 days, implants harvested for adipose tissue formation were cut in half. The specimens for H&E, Toluidine Blue and Masson staining were dehydrated and embedded in paraffin, cut into 5 μm sections and stained. Specimens for Oil red O staining were embedded in Tissue-Tek OCT medium, cut into sections 7 mm thick, and stained. Bone tissues were soaked in 0.5 mol/L EDTA for decalcification, sectioned and subjected to H&E staining as described above. Histological sections were observed and images were captured using a microscope.
Micro-computed tomography (micro-CT)
Images of femurs were obtained at a current of 220 μA, voltage of 60 kV, and exposure time of 1 500 ms. Images of cranial bones were obtained at a current of 500 μA, voltage of 80 kV, and exposure time of 1 500 ms using the Inveon MM system (Siemens, Munich, Germany). Inveon Research Workplace (Siemens) was used to calculate the BV/TV, Tb. Sp, Tb. N, and BMD independently in triplicate. The investigators were blinded to the treatment allocation.
RNA sequencing analysis
Total RNA (1 μg per sample) was used to construct a miRNA library. After purification of small RNA from total RNA and reverse transcription, the final products (100–120 bp) were sequenced on the BGISEQ-500 platform (BGI-Shenzhen, China). Clean tags were generated using SOAPnuke v. 1.5.2 software (BGI-Shenzhen, China). These tags were mapped to the reference genome and several sRNA databases using Bowtie257. For mapping to the Rfam database v. 13.0, cmsearch58 was used. Gene expression levels were quantified by counting the absolute numbers of molecules identified by the unique molecular identifiers59. Data processing and enrichment analysis were conducted using R v. 3.1.1 software. Differential expression analysis was performed using the DEGSeq260.
Statistical analysis
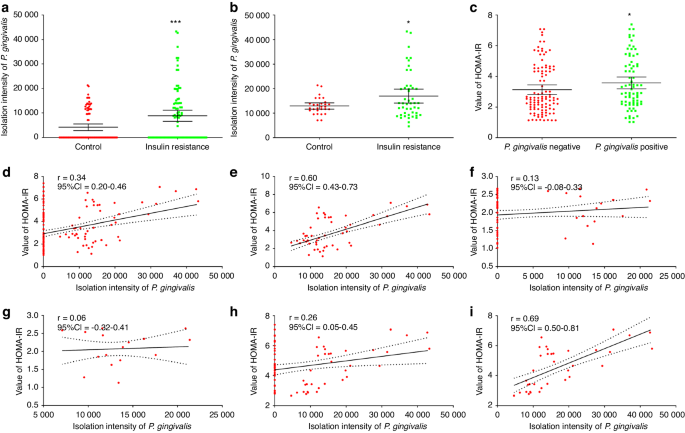
Data are shown as means ± standard deviations. Statistical analysis was performed using Prism software (GraphPad Software, Inc., La Jolla, CA). Between-group comparisons were performed using independent two-tailed Student’s t-tests, and multiple-group comparisons were conducted using one-way ANOVA followed by a Tukey’s post hoc tests. A two-tailed value of P < 0.05 was considered indicative of statistical significance.




留言 (0)