
記住我



Through RNA-Seq analysis of HNSCC samples (3 tumors and 3 para-carcinoma tissues), we identified 8 pivotal marker genes (PCDH7, CDH2, ITGA3, SEMA7A, TMEM132A, CD276, TMEM2, GPR158) that were overexpressed in tumor specimens (Fig. 1a, Supplementary Table 1). Subsequently, high enrichment of these molecules was further verified in 5 HNSCC cell lines, along with the finding that SEMA7A presented marked upregulation in all cell types (Fig. 1b, c), indicating its participatory role in tumor progression. However, the underlying mechanism of SEMA7A in HNSCC is poorly defined.
Fig. 1
Identification of the promotive role of SEMA7A in HNSCC progression. a Distinct transcriptive level of SEMA7A between HNSCC tumors and adjacent normal tissues in HNSCC-TCGA database. b Verification of the transcriptive divergence in clinical specimens of HNSCC (20 para-tumors vs. 20 tumors). c The representative IHC images with diverse intensities (negative, low, intermediate and high) staining in tissue microarray from HNSCC tumors. d Kaplan–Meier plots of the overall survival of HNSCC patients stratified by the IHC score of SEMA7A (p value was calculated using the log-rank test). e, f Expression of SEMA7A protein in five representative HNSCC cell lines by immunoblot (e) and semi-quantitative analysis (f). g, h Cell proliferation measurement in HN6 (g) and HN30 (h) cell lines transfected with scramble and SEMA7A-shRNA, respectively. i–k. Confirmation of SEMA7A knockdown-mediated tumor growth inhibition in subcutaneous xenograft model (n = 4) presented as general review (i), tumor weight (j) and growth curve (k). (Data were shown as mean ± SEM, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.000 1)
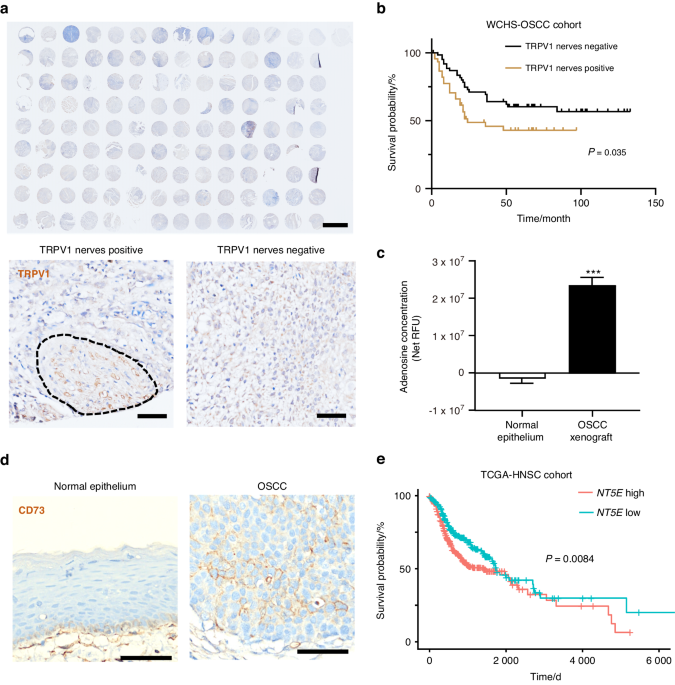
SEMA7A is widely expressed in human tissues and organs, such as the vitreous humor, colonic epithelial cells, and testis (Fig. 2a). In addition, SEMA7A is highly expressed across different cancer types (for example, HNSCC), with overexpression in tumor tissues compared to paired para-cancerous samples (Fig. 2b). Significant upregulation of SEMA7A in the oral cavity, oropharynx and hypopharynx was also discovered by analysis of the TCGA-HNSCC database (Figs. 1a, 2c). This was further validated by polymerase chain reaction (PCR) analyses in fresh HNSCC tumor tissues and precancerous lesions (20 vs. 20) (Fig. 1b). A high abundance of SEMA7A in tumors indicated a poorer clinical outcome of overall survival (OS) in the TCGA database (Fig. 2d). Ultimately, tissue microarray analysis (TMA) with 63 HNSCC samples confirmed the positive correlation of higher expression of SEMA7A in tumor cells (high: positive ratio ≥ 20%; low: positive ratio <20%) with poorer prognosis (Fig. 1c, d).
Fig. 2
Aberrant N-glycosylation of SEMA7A in HNSCC cell lines. a Western blot analysis of SEMA7A (glycosylated and non-glycosylated form) in four HNSCC cell lines pre-treated with or without PNGase F (+N) and O-Glycosidase (+O). b Western blot analysis of SEMA7A in HNSCC cell lines pre-incubated with tunicamycin and swainsonine at the designed timepoint of 24 and 48 h. c Flow cytometry measuring of different lectins (VVL, SNA, PHA-L, Con A and LCA) on the membrane of HN6 (left) and HN30 (right) cells. d Schematic diagram of mutation plasmids construction based on the five glycosylation sites of SEMA7A by substituting of asparagines (N) to glutamine (Q). e Verification of the exogenous expression and deglycosylated statues of wild-type SEMA7A or the single-site N-glycosylation SEMA7A mutant in HN6 (left) and HN30 (right) cells transfected with SEMA7A-shRNA. f Comparison of the deglycosylated efficiency of tunicamycin with SEMA7A-3NQ and SEMA7A-5NQ mutants in HN6 (left) and HN30 (right) cells. g Comparison of the deglycosylated statue in HN6 cells treated with PNGase F (+N), O-Glycosidase (+O) and SEMA7A-5NQ mutation plasmid
To explore the oncogenic potential of SEMA7A in HNSCC, we carried out in vitro and in vivo verification analyses. Through protein and mRNA level measurement, we finally selected the HN6 and HN30 cell lines as the major objects for further investigation (Figs. 1e, f, 2e). Via a knockdown strategy, we established stable clones with SEMA7A knockdown with ideal knockdown efficiency in HN6 and HN30 cell lines by using a lentiviral vector. In vitro cell proliferation experiments demonstrated an obviously lower proliferation rate in SEMA7A knockdown clones (Fig. 1g, h). To further confirm this growth inhibition in vivo, HN6 knockdown cells were injected into the flank region of BALB/c nude mice, and downregulation of SEMA7A was found to significantly suppress the tumorigenesis of HN6 cells in vivo, as evidenced by the tumor mass and growth curve (Fig. 1i–k). Collectively, the current data support the concept that SEMA7A is an oncoprotein in HNSCC.
Evaluating the glycosylation of SEMA7A in HNSCCWhen detecting the protein expression of SEMA7A in different HNSCC cell lines, we found a dramatic band shift toward a higher molecular weight in tumor cells compared with normal epithelia (Fig. 1e), suggesting the presence of posttranslational modifications, especially glycosylation, which is intimately correlated with the molecular weight of SEMA7A. Incubation with peptide-N-glycosidase F (PNGase F) to remove N-glycosylation (deglycosylation) demonstrated band separation to a nonglycosylated form, which was not observed after treatment with O-glycosidase to remove O-glycans (Fig. 2a). Subsequently, incubation of tumor cells with tunicamycin, a well-known inhibitor of N-glycosylation, also showed results similar to those observed after PNGase F treatment; however, this protein band shift was not detected in swainsonine incubated group (Fig. 2b). As mentioned above, this band shift may be ascribed to the aberrant N-glycosylation of SEMA7A in cancer cells. To determine the specific patterns of N-glycosylation, we performed flow cytometry by using anti-lectin antibodies (Vicia villosa lectin, VVL; Sambucus nigra lectin, SNA; Phaseolus vulgaris leucoagglutinin, PHA-L; Concanavalin A, Con A; Lens culinaris agglutinin, LCA) and revealed that LCA was the dominant form of SEMA7A N-glycosylation in both HN6 and HN30 cells (Fig. 2c).
To delineate the molecular mechanism by which aberrant glycosylation of SEMA7A promotes HNSCC oncogenesis, we conducted glycosylation intervention experiments by constructing mutant plasmids. Through amino acid sequence analysis, five NXT motif sites were found in human SEMA7A (Asn 105, 157, 258, 330, and 602) (Fig. 2d). We then generated the SEMA7A-WT and SEMA7A-5NQ mutants by substituting asparagine (N) with glutamine (Q). The effect of deglycosylation was validated in SEMA7A-shRNA cell models (Fig. 2e). Samples containing the N105Q, N157Q, and N258Q proteins showed a slight band shift that was not found in cells with N258Q and N602Q. To elucidate whether the band shift in 5NQ was attributed to glycosylation of residues 105, 157, and 258, we reconstructed the 3NQ mutant plasmid and found that all mutations in glycosylated sites contributed to deglycosylation (Fig. 2f). Compared with the deglycosylation efficiency of tunicamycin and PNGase F treatment, the 5NQ mutation completely ablated SEMA7A N-glycosylation, as indicated by the identical band shift (Fig. 2g).
Glycosylation status determined the protein degradation patternsGlycosylation has been reported to participate in protein degradation and stability. After treatment with the protein synthesis inhibitor cycloheximide (CHX), endogenous glycosylated SEMA7A exhibited a higher turnover rate than the SEMA7A protein with deglycosylation induced by tunicamycin (Fig. 3a). This difference in degradation rate was further confirmed by evaluation of exogenous SEMA7A-WT and SEMA7A-5NQ in the presence of CHX (Fig. 3a, b). These data indicated a more stable status of nonglycosylated SEMA7A, with less susceptibility to degradation. Subsequently, we investigated the underlying mechanism to interpret this biological phenomenon. Under nonreducing conditions, which had no significant influence on that of SEMA7A-WT and SEMA7A-3NQ, SEMA7A-5NQ lost the capability to form protein dimers, implying dysfunction in SEMA7A-5NQ signal transduction (Fig. 3c). Protein degradation is accomplished through three main pathways: (1) the ubiquitin‒proteasome system (UPS); (2) autophagic degradation; and (3) endoplasmic reticulum-associated degradation (ERAD). By incubation with inhibitors (MG132 for the UPS, hydroxychloroquine (HCQ) for autophagy and bortezomib (BTZ) for ERAD), we found completely distinct degradation pathways between nonglycosylated (through ERAD) and glycosylated (via the UPS) SEMA7A, as evidenced by the different protein accumulation patterns observed following treatment with the degradation inhibitors (Fig. 3d). This degradation trend was further proven by immunoblot analysis of exogenous Flag-tagged SEMA7A proteins at different timepoints of 0, 6 and 12 h (Fig. 3e). To further verify the difference in UPS-dependent proteolysis, cells were sequentially transfected with ubiquitin and SEMA7A-WT/5NQ and then incubated with tunicamycin and MG132. The data showed that the UPS system failed to facilitate the degradation of ubiquitinated SEMA7A-5NQ (Fig. 3b). Conversely, cells expressing SEMA7A-5NQ demonstrated relatively lower activity of enzymes involved in ERAD, such as OS9 and ERECL1, which indicated that ERAD-dependent degradation was suppressed when SEMA7A was deglycosylated, thus leading to relatively higher protein stability (Fig. 3c). We also speculated that the secreted form of SEMA7A is correlated with this difference in degradation. However, no difference was found between SEMA7A-WT- and 5NQ-overexpressing cells by using ELISA (Fig. 3d). Thus, the more stable state of nonglycosylated SEMA7A may contribute to the lower activity of ERAD enzymes.
Fig. 3
Distinct statues of glycosylation determined the SEMA7A protein degradation manner. a, b The intensity of endogenous (a) and exogenous (b) SEMA7A protein in HN6 (left) and HN30 (right) cells pre-treated with CHX (20 μmol/L) at designed time intervals in the presence of 2.5 μg/mL tunicamycin or not. c Detection of SEMA7A protein dimer formation in HN6 (up) and HN30 (down) cells transfected with different mutation plasmids at reducing and non-reducing conditions. d Western blot analysis of SEMA7A in HN6 (up) and HN30 (down) cells exogenously expressing either wild type (left) or 5NQ mutant (right) in the presence of CHX (20 μmol/L for 12 h) and subsequent HCQ, MG132 and BTZ for 6 h. e Protein expression of exogenous Flag-tag in HNSCC cells treated with CHX, and followed by HCQ, MG132, and BTZ at indicated time intervals
N-glycosylation of SEMA7A promoted HNSCC progressionTo clarify whether glycosylation of SEMA7A is indispensable for HNSCC oncogenesis, in vitro and in vivo functional alterations were explored. The subcellular distribution is a pivotal factor in the ability of proteins to execute their precise biological functions. N-glycosylation was necessary for SEMA7A trafficking from the cytoplasm to the cytomembrane, as evidenced by the increased colocalization of SEMA7A-5NQ with endoplasmic reticulum (calreticulin) and Golgi (GM130) markers compared to that of SEMA7A-WT observed by laser confocal microscopy (LCFM), indicating retention of the SEMA7A-5NQ molecule in the cytoplasm (Fig. 4a). The contribution of individual N-glycosylation sites to SEMA7A subcellular localization was also determined by LCFM, with the finding that none of the single-site mutations had an influence on its cytomembrane trafficking (Fig. 4). Removal of N-glycosylation enhanced the detection of SEMA7A, as proven by the significant increase in the fluorescence intensity of SEMA7A in both the HN6 and HN30 cytomembranes compared with that in the control and O-glycan removal treatment groups (Fig. 4b). Plexin C1 is a classical receptor with high affinity for SEMA7A.30,31 We then explored whether deglycosylation of SEMA7A affected the formation of the SEMA7A/Plexin C1 complex. SEMA7A-WT/5NQ cells were incubated with recombinant Plexin C1 protein (50 µg/ml), and the affinity difference was assessed by flow cytometry. Unsurprisingly, complete ablation of N-glycosylation significantly attenuated the binding of SEMA7A to Plexin C1 (Fig. 4c).
Fig. 4
Aberrant glycosylation of SEMA7A facilitated HNSCC progression. a Confocal microscopy images of co-distribution of SEMA7A with Endoplasmic Reticulum (calreticulin, left) and Golgi markers (GM130, right) in HNSCC cells transfected with WT (up) and 5NQ (down) plasmids. b Immunofluorescence observation of SEMA7A distribution and intensity in HN6 (up) and HN30 (down) cells pre-treated with PNGase F and O-Glycosidase. c Flow cytometry analysis of the binding activity between SEMA7A-WT/ Plexin C1 and SEMA7A-5NQ/ Plexin C1 in HN6 (up) and HN30 (down) cells. d, e Diverse cellular proliferation rates of HN6 (d) and HN30 (e) cells transfected with various mutation plasmids detecting by CCK-8. f–h Tumor growth of SEMA7A-WT and 5NQ HN6 cells in BALB/c nude mice, showing as gross view of isolated tumors (f), tumor mass (g), and growth curve (h). Data was shown as the mean ± SD (n = 5), P value was calculated by Student’s t test (****P < 0.000 1)
Whether impairment of SEMA7A N-glycosylation blocks tumor progression is also a pivotal issue. In vitro proliferation analysis demonstrated that deglycosylation of SEMA7A dramatically decreased cell viability and was negatively correlated with tumor invasion in both HN6 and HN30 cells (Figs. 4d, e, 5). These results were further confirmed in an in vivo mouse model that showed a decreased tumor mass and volume (Fig. 4f–h). Taken together, these data demonstrated an important role of SEMA7A N-glycosylation in tumorigenesis.
Fig. 5
Identification of FUT8 as glycotransferases for SEMA7A aberrant glycosylation. a qRT-PCR analysis of 16 N-glycosyltransferases mRNA expression in HNSCC cell lines (HN4, HN6, and HN30). b Lectin pull-down analysis (LCA, Con A, PHA-L, SNA) of SEMA7A in HNSCC cells transfected with different si-RNAs filtered from 16 N-glycosyltransferases mRNA expression. c Representative confocal images of the colocalization of FUT8 with Endoplasmic Reticulum (calreticulin) and Golgi markers (GM130, Giantin, and TGN46) in HN6 and HN30 cells. Scale bar, 10 μm. d Immunofluorescence co-distribution of SEMA7A with LCA lectin in HN6 (up) and HN30 (down) cells treated with si-NC, si-FUT8 and 2F-Fuc. Scale bar, 10 μm. e Flow cytometry measuring the binding activity of SEMA7A with Plexin C1 in HNSCC cells transfected with si-NC and si-FUT8, followed by SEMA7A-WT and 5NQ. f Cell viability detection of HN6 (up) and HN30 (down) cells transfected with si-NC and si-FUT8 at designed timepoints (0, 1, and 3 days). g Representative IHC images with diverse FUT8 intensities (negative, low, and high) staining in tissue microarray from HNSCC tumors. Scale bar, 50 μm. h Kaplan–Meier plots of the overall survival of HNSCC patients stratified by the IHC score of FUT8 (p value was calculated using the log-rank test). (Data were shown as mean ± SEM, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.000 1)
Fucosyltransferase FUT8 catalyzes aberrant SEMA7A N-glycosylationSubsequently, we sought to identify the underlying mechanism that regulates the aberrant N-glycosylation of SEMA7A. The genes encoding sixteen glycotransferases responsible for N-glycosylation were obtained, and their transcript levels were analyzed in different HNSCC cell lines.11 Among these genes, FUT8, ST3GLA1, B3GAT1, B3GAT2, GCNT3, and MAGT3 were significantly upregulated in both HN6 and HN30 cells compared with normal epithelia (Fig. 5a). Based on this, we then validated the determined role of these six glycotransferases in the N-glycosylation of SEMA7A by using lectin blotting. The successful synthesis of the siRNAs and their knockdown efficiency in both cell lines were verified (Fig. 6). First, knockdown of these glycotransferases did not affect the total protein expression level of SEMA7A. By using LCA, Con A and SNA lectin pull-down assays, we found an obvious reduction in SEMA7A enrichment in si-FUT8, si-ST3GLA1, and si-GCNT3 cells. Regarding PHA-L lectin enrichment, a decrease in SEMA7A enrichment was found in si-FUT8, si-B3GAT1, si-B3GAT2, and si-GCNT3 cells. LCA, representing core fucosylation, was the most abundant lectin in both HN6 and HN30 calls, as determined by flow cytometry (Fig. 5b). Therefore, we speculated that FUT8 may be the major catalyzer responsible for the aberrant glycosylation of SEMA7A.
Fig. 6
FUT8 mediated abnormal glycosylation of SEMA7A through FUT8-SEMA7A proteins interaction. a Total (Input) or immunoprecipitated (IP) lysates from HN6 cells transfected with Flag-tagged SEMA7A-WT together with HA-tagged FUT8-WT or FUT8-R365A were immunoblotted using the indicated antibodies. b IP analysis of the interaction of exogenous SEMA7A-WT-Flag with FUT8-WT-HA in the presence or absence of tunicamycin for 24 h. c LCA affinity of whole-cell lysate of HN6 cells overexpressed with SEMA7A-WT-Flag as well as FUT8-WT-HA or FUT8-R365A-HA by western blot with anti-SEMA7A. d Schematic diagram of truncated fragments of SEMA7A and FUT8 according to the different functional domains. e, f HN6 cells were transiently transfected with truncated HA-tagged FUT8 (e) and Flag-tagged SEMA7A (f) mutations, followed by immunoprecipitation with anti-HA and Flag beads and subsequent immunoblot analysis with anti-Flag and HA
FUT8 is mainly distributed in the Golgi, as evidenced by the colocalization of the FUT8 protein with Golgi markers (GM130 for the cis-Golgi, Giantin for intercisternal cross-bridges of the Golgi complex, and TGN46 for the trans-Golgi) but not the ER marker calreticulin (Fig. 5c). Downregulation of FUT8 had no significant influence on the transcript and protein levels of SEMA7A (Fig. 7). Subsequently, we investigated whether FUT8 knockdown has a determining role in the core fucosylation of SEMA7A. 2F-Fuc is a typical inhibitor of core fucosylation and significantly abolishes LCA binding to SEMA7A. Similarly, FUT8 knockdown also dramatically decreased the fluorescence intensity of LCA, indicating that FUT8 is necessary for the core fucosylation of SEMA7A (Fig. 5d). In addition, downregulation of FUT8 markedly attenuated the binding of SEMA7A-WT to Plexin C1 (si-NC/SEMA7A-WT, 21 713 ± 940; si-FUT8/SEMA7A-WT, 16 651 ± 1108). However, this alteration in binding was not observed for SEMA7A-5NQ/Plexin C1 (si-NC/SEMA7A-5NQ, 11 252 ± 894; si-FUT8/SEMA7A-5NQ, 11 066 ± 688) (Figs. 5e, 8). Then, we tried to clarify whether overexpression of FUT8 in tumors promotes oncogenesis. Knockdown of FUT8 in tumor cells significantly abrogated in vitro proliferation (Fig. 5f). Through HNSCC tissue microarray analysis, we further confirmed that higher expression of FUT8 in tumor cells (high: positive ratio ≥ 10%; low: positive ratio < 10%) was intimately associated with poorer prognosis (Fig. 5g, h).
Fig. 7
EGF and TGF-β1 signaling induce N-glycosylation of SEMA7A. a, b GO and KEGG analyses of the HNSCC tumors from TCGA-HNSCC stratified according to the different abundance of SEMA7A. c, d Western blot analysis of the binding affinity of SEMA7A with LCA in HN6 (c) and HN30 (d) cells treated with EGF, IGF, FGF, HGF, and TGF-β1 for overnight. e Glycosylation recovery of HN6 cells expressing with wild type SEMA7A or its N-glycosylation site mutants in the presence or absence of TGF-β1 and EGF. f, g The interaction activity of exogenous SEMA7A-WT-Flag (f) and SEMA7A-5NQ-Flag (g) with FUT8-WT-HA in the presence or absence of TGF-β1 and EGF by using IP and subsequent western blot analysis. h The influence of FUT8 intervention on the EGF and TGF-β1 signaling mediated N-glycosylation of SEMA7A by using lectin enrichment and western blot. i The determined role of EMT and MET events on the binding affinity of LCA with SEMA7A in lectin blot
Fig. 8
Deglycosylation of SEMA7A contributes to the EGFR targeted and immune therapy. a, b Correlation analysis of SEMA7A abundance with tumor infiltrate lymphocytes (a) and checkpoint expression (b). c The biological influence of SEMA7A N-glycosylation on PD-L1 protein expression in membrane, cytoplasm and nucleus using cell fractionation and subsequent immunoblot analysis. d The indicated HN6-SEMA7A KD cells were transiently transfected with SEMA7A-WT and 5NQ plasmids in the presence of tunicamycin or not, then cocultured with CD3/CD28-activated human CD8+ T-lymphocyte cells. Representative plots of the tumor apoptosis were measured by FACS. e Detection of the checkpoints (PD-1, CTLA4, TIM3, and LAG3) expression in human CD8+ T-lymphocyte cells cocultured with tumor cells transfected with SEMA7A-WT and 5NQ in the presence or absence of tunicamycin by flow cytometry. f Elisa measuring of IL-2 release in Jurkat cells cocultured with HNSCC cells overexpressed with SEMA7A-WT and 5NQ or pre-treated with tunicamycin. g The therapeutic protocol of tumor growth of Sema7a-WT re-expressed SCC7-Sema7a-KD cells in C3H mice following treatment with 2F-Fuc and anti-PD-L1 antibody. h, i In vivo tumor growth in four groups with different treatments presented as tumor specimens (h) and growth curve (i). (Data were shown as mean ± SEM, ** P ≤ 0.01, *** P ≤ 0.001, ****P ≤ 0.000 1)
Interaction of FUT8 with SEMA7A during aberrant glycosylationWe speculated that FUT8 may directly interact with SEMA7A during glycan modification. WT-HA-tag FUT8 and R365A-HA-tag FUT8 (inactive mutant) plasmids were constructed and co-transfected with the SEMA7A-WT-Flag-tag plasmid into tumor cells. Immunoprecipitation (IP) of HA verified the direct binding of FUT8 to glycosylated SEMA7A. However, FUT8-R365A showed decreased binding capacity compared with FUT8-WT, indicating that the active form of FUT8 was indispensable for its catalyzing role in glycosylation (Fig. 6a). Ultimately, we tried to identify whether the glycosylation status of SEMA7A was necessary for its protein‒protein interaction (PPI). In the presence of tunicamycin, deglycosylated SEMA7A exhibited a greater binding ability than the glycosylated form (Fig. 6b). As shown above, eradication of glycosylation may enhance antigen exposure and detection, which may account for the increased interaction between FUT8 and deglycosylated SEMA7A. To delineate whether the functional status of FUT8 was associated with SEMA7A glycosylation, we evaluated LCA enrichment, which showed a significant reduction in the binding of LCA to SEMA7A in FUT8-R365A cells compared with FUT8-WT cells, indicating that the active form of FUT8 was necessary for glycan modification of SEMA7A (Fig. 6c).
STX18 is a SNARE protein that is involved in membrane trafficking between the ER and Golgi.32 Through PPI network analysis, we found that STX18 is a candidate binding partner for FUT8. This finding led us to speculate that STX18 may act as a trafficking protein during FUT8-mediated SEMA7A glycosylation. STX18 knockdown has no determined role in SEMA7A protein expression and glycosylation (Fig. 9a). By co-transfection with Myc-tag STX18, HA-tag FUT8, and Flag-tag SEMA7A, co-IP verified pairwise interactions among the three proteins (Fig. 9b). These data indicated that STX18 is a transport protein for the FUT8/SEMA7A complex. To further investigate the influence of protein synthesis and transport on the pairwise interactions of these three proteins, co-transfected tumor cells were incubated with CHX (inhibitor of protein synthesis) and brefeldin A (BFA, inhibitor of protein trafficking), with the finding that BFA significantly abrogated the interactions between STX18 and SEMA7A, FUT8 and STX18 and between FUT8 and SEMA7A. However, inhibition of protein synthesis only affected the binding of STX18 and SEMA7A (Fig. 9c–e). These Co-IP data demonstrated that protein transport is a necessary process for FUT8-mediated SEMA7A glycosylation.
Fig. 9
N-glycosylation of SEMA7A induced RBM4 upregulation is associated with PD-L1 alternative splicing. a, b Proteomic analysis of the protein lysates from HN6 cells overexpressed with SEMA7A-WT and 5NQ (n = 3). Functional enrichment of differential expressed proteins between two groups were subjected to KEGG pathway analysis. c In vitro proliferation of si-NC/si-RBM4 (left) and Vector/RBM4-OE plasmids (right) transfected HNSCC cells using CCK8 assay at indicated time interval. d Cell proliferation measuring of HNSCC cells sequentially transfected with SEMA7A-WT/5NQ and si-NC/si-RBM4. e Detection of PD-L1 alternative splicing in HNSCC cells following RBM4 intervention. f Expression of PD-L1 isoforms in HN6 cells transfected with SEMA7A-WT and 5NQ using real-time PCR. g Transcriptive level of PD-L1 isoforms in HN6 cells sequentially transfected with SEMA7A- 5NQ and si-NC/si-RBM4. (Data were shown as mean ± SEM, *P ≤ 0.05,**P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.000 1)
SEMA7A is composed of SPI, Sema, PSI and IG-like domains, and FUT8 includes TM, glycosyltransferase and SH3 domains.33,34 To pinpoint the specific interaction domain between FUT8 and SEMA7A, we generated 4 Flag-tagged SEMA7A truncation fragments and 3 HA-tagged FUT8 truncation fragments (Fig. 6d). HN6 cells were cotransfected with SEMA7A full-length/4 fragments with FUT8-WT or with FUT8 full-length/4 fragments with SEMA7A-WT and subsequently analyzed by Flag or HA enrichment and further immunoblot analysis. A missing band was found for the fragment with deletion of the Sema domain after HA enrichment, and bands were absent in the Δ Glycosy and Δ SH3 fragments following anti-Flag precipitation (Fig. 6e, f). These data supported the concept that the Sema domain in SEMA7A and the glycosyltransferase/SH3 domains in FUT8 are the specific domains necessary for this protein‒protein interaction. The subcellular distribution of the truncated FUT8 fragments, as evaluated by confocal microscopy, further verified the glycosyltransferase/SH3 domain specificity, as evidenced by the different localization statuses of the truncations with ER and Golgi markers (Fig. 10a). In addition, the recognition of SEMA7A by Plexin C1 was distinct from that of SEMA7A by FUT8. Flow cytometry showed that the Sema, PSI and IG-like domains are all indispensable for the formation of the SEMA7A/Plexin C1 complex (Fig. 10b).
Fig. 10
Molecular mechanism of FUT8 mediated aberrant SEMA7A glycosylation and its promotive role in HNSCC progression
EGF and TGF-β1 induce SEMA7A glycosylation via different mechanismsThrough GO and KEGG analyses of the HNSCC database from the cancer genome atlas (TCGA-HNSCC), we discovered that a high abundance of SEMA7A was associated with extracellular matrix (ECM) organization and ECM receptor interaction, indicating that SEMA7A participated in tumor microenvironment remodeling (Fig. 7a, b). To identify the ECM-related cytokines that govern SEMA7A N-glycosylation, we treated HNSCC cell lines with several growth factors, including transforming growth factor (TGF-β1), epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF) and hepatocyte growth factor (FGF). After LCA enrichment, dramatic accumulation of SEMA7A was found only in TGF-β1- and EGF-treated HN6 and HN30 cells, suggesting the inducing effect of these two cytokines on the N-glycosylation of SEMA7A (Fig. 7c, d). Therefore, we investigated whether TGF-β1 and EGF mediate SEMA7A glycosylation via a similar molecular mechanism. First, we examined the reglycosylation of individual mutation sites following growth factor incubation and found that compared with TGF-β1 treatment, EGF treatment resulted in an increased abundance of SEMA7A glycosylated at residues 105 and 602 (Fig. 7e). Next, we investigated the determining role of growth factors in the interaction between SEMA7A and FUT8. Co-IP analysis showed that compared with TGF-β1, EGF significantly increased the binding activity of SEMA7A-WT toward FUT8, an effect that failed to be repeated for SEMA7A-5NQ (Fig. 7f, g). Furthermore, we tried to elucidate whether FUT8 intervention has a similar effect on TGF-β1- and EGF-induced SEMA7A glycosylation. HN6 cells were treated with si-NC and si-FUT8 prior to incubation with growth factors, and the binding of the SEMA7A protein with LCA then was detected. Interestingly, FUT8 knockdown substantially attenuated the core fucosylation of SEMA7A induced by TGF-β1 but not that induced by EGF (Fig. 7h).
Subsequently, we sought to delineate the association of SEMA7A glycosylation with TGF-β1-induced epithelial-mesenchymal transition (EMT). Through EMT and mesenchymal-epithelial transition (MET) model establishment (Supplementary Fig. 11a), we determined that the binding of LCA to SEMA7A was altered in tandem with the EMT and MET processes (Fig. 7i). Furthermore, during MET, FUT8 overexpression alleviated the deglycosylation of SEMA7A (Fig. 7i). These findings indicated that TGF-β1-mediated SEMA7A glycosylation was regulated by FUT8 in the EMT process. We next investigated the role of SEMA7A and FUT8 in EMT through siRNA and plasmid introduction followed by TGF-β1 stimulation. Surprisingly, the alteration of SEMA7A glycosylation and FUT8 expression had no obvious influence on the EMT process (Supplementary Fig. 11b, c). These results indicated that FUT8-mediated SEMA7A glycosylation was regulated by EMT but not involved in the regulation of EMT. Taken together, these findings indicated that TGF-β1 mediates SEMA7A glycosylation mainly through EMT in a manner mediated by FUT8, while EGF mediates glycosylation mainly by increasing the binding activity of SEMA7A toward FUT8.
N-glycosylation of SEMA7A mediates the efficacy of EGFR targeting and immunotherapyEGF signaling is one of the main inducers of SEMA7A glycosylation, and this observation prompted us to hypothesize that inhibition of SEMA7A glycosylation may enhance the efficacy of EGFR-targeted therapy. To this end, we first tested the activation of EGFR signaling in SEMA7A-WT/5NQ cells following EGF incubation. The phosphorylation of ERK1/2, STAT3 and EGFR was dramatically attenuated in SEMA7A-5NQ cells, indicating that SEMA7A glycosylation has a determining role in the transduction of EGFR signaling (Supplementary Fig. 12a). Subsequently, HNSCC cell lines were treated with EGFR inhibitors (gefitinib and erlotinib), and SEMA7A-5NQ cells were found to be more sensitive to EGFR-targeted therapy, as evidenced by the elevated cleaved Caspase-3 activity and decreased cell proliferation rate (Supplementary Fig. 12b, c).
We further investigated the association of SEMA7A with the immunosuppressive microenvironment. Through TCGA-HNSCC data analysis, we proved that overexpression of SEMA7A was positively correlated with elevation of immune checkpoint (PDCD1LG2, PD-L1, CTLA4, TIM3, TIGIT, and PD-1) expression, a decrease in activated CD8+ T cells and increased infiltration of exhausted CD8+ T cells (Fig. 8a, b). Inspired by these findings, we detected PD-L1 expression in tumor cells by cell fractionation and found that deglycosylation of SEMA7A significantly reduced the PD-L1 levels in the cytoplasm and nucleus but not the cytomembrane (Fig. 8c). Subsequently, we isolated CD8+ T cells from peripheral blood and then cocultured them with SEMA7A-WT/5NQ tumor cells and tunicamycin. Flow cytometry showed a higher proportion of apoptotic cells in the SEMA7A-5NQ group, an observation identical to that in tunicamycin-treated cells, indicating that cells with deglycosylation of SEMA7A were more susceptible to CD8+ T cell-mediated cytotoxicity (Fig. 8d). Next, we investigated the immunosuppressive effect of SEMA7A glycosylation on CD8+ T cells. Aberrant glycosylation of SEMA7A resulted in a higher proportion of exhausted CD8+ T cells than did SEMA7A-5NQ, as characterized by the distinct increases in the PD-1, LAG3, CTLA4, and TIM3 levels in T cells (Fig. 8e). In addition, deglycosylation of SEMA7A may partially rescue the cytotoxicity of Jurkat cells, as evidenced by increased IL-2 secretion (Fig. 8f). Finally, we investigated the antitumor efficiency of 2-fluoro-L-fucose (2F-Fuc, a classical inhibitor of cellular core fucosylation) treatment combined with anti-PD-L1 immunotherapy in C3H (immunocompetent) mice, attempting to verify whether blocking the core fucosylation of SEMA7A with 2F-Fuc could improve the outcome of immunotherapy. In C3H mice bearing Sema7a-5NQ-re-expressing SCC7-Sema7a-KO xenograft tumors, there was no combinatorial effect when the tumors were treated with 2F-Fuc and anti-PD-L1 antibodies (data not shown). However, in C3H mice bearing Sema7a -WT-re-expressing SCC7- Sema7a-KO xenograft tumors, the combined treatment with 2F-Fuc and anti-PD-L1 antibodies conspicuously suppressed tumor growth and expansion, as confirmed by the tumor observation and growth curves of the xenograft tumors (Fig. 8g–i). Taken together, these findings indicated that inhibition of SEMA7A glycosylation enhances the efficacy of T-cell-mediated immunotherapy.
Here, we also evaluated the correlation of SEMA7A glycosylation with immunogenic cell death (ICD), which can be induced by doxorubicin (DOX). The typical features of ICD are increased calreticulin (CRT) translocation, elevated autophagy and decreased nuclear HMGB1. In our data, SEMA7A-5NQ and tunicamycin significantly augmented CRT expression and attenuated HMGB1 nuclear accumulation (Supplementary Fig. 13a, b). Moreover, SEMA7A-5NQ cells were more sensitive to DOX therapy, as evidenced by the decreased cell viability (Supplementary Fig. S13c). These results suggested that deglycosylation of SEMA7A markedly enhanced DOX’s activity in ICD induction.
RBM4 upregulation mediated by N-glycosylation of SEMA7A is associated with PD-L1 alternative splicingWe finally sought to explore downstream signaling in cells with different statuses of SEMA7A N-glycosylation. After transfection with SEMA7A-WT/5NQ, HN6 cells were then subjected to protein extraction and subsequent mass spectrometry and proteomics analysis. KEGG pathway analysis on the basis of differentially expressed proteins (DEPs) showed that the spliceosome ranked at the top in both the mass spectrometry and proteomics data (Fig. 9a, b). By filtering of the list of spliceosome-related DEPs, we found an interesting protein, RBM4, RNA-binding motif 4, a splicing regulator mediating alternative splicing programs (Supplementary Table 2).35,36,37 The elevation of RBM4 expression induced by SEMA7A-5NQ was further verified in HN30 cells (Supplementary Fig. 14a). We then tried to uncover the molecular mechanism of SEMA7A-5NQ-triggered upregulation of RBM4. Compared to that in the control group, SEMA7A-WT inhibited RBM4 transcription, while SEMA7A-5NQ partially rescued the mRNA expression of this splicing factor (Supplementary Fig. 14b). In addition, we observed a lower turnover rate of the RBM4 protein in SEMA7A-5NQ-transfected cells at 0, 4, 12, and 24 h than in SEMA7A-WT cells (Supplementary Fig. 14c). All of these results indicated that SEMA7A-5NQ upregulated RBM4 expression by augmenting its mRNA transcription and retarding its protein degradation.
RBM4 has been reported to be involved in cell proliferation. To delineate the functional role of RBM4 in the tumor microenvironment, we performed gain and loss of function experiments. In vitro cell viability analysis demonstrated that RBM4 has a negative regulatory role in HNSCC cells (Fig. 9c). Then, we sought to determine whether the SEMA7A-5NQ-mediated cell growth inhibition was regulated by RBM4. Tumor cells were cotransfected with si-RBM4/RBM4-OE and SEMA7A-WT/5NQ and were then subjected to a CCK8 assay. RBM4 was the main regula
留言 (0)