
記住我


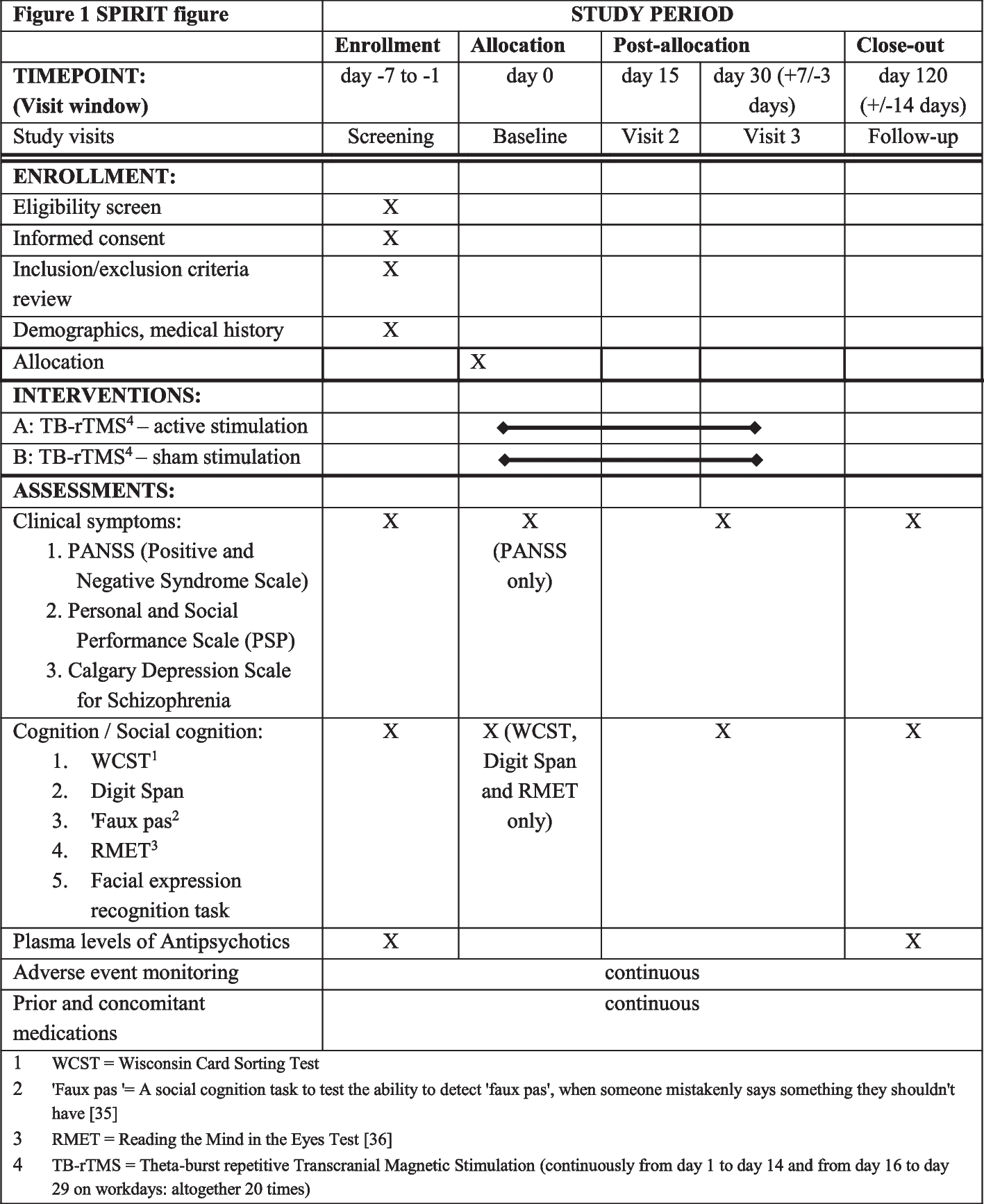

The reporting of this protocol is based on the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT), which is found in Additional file 2 with SPIRIT checklist [27].
ObjectivesThe primary objective of the INTERCEPT study is to evaluate the efficacy of addition of TCZ to SOC as compared to SOC alone in reducing the decline of graft function from baseline at 24 months after start of treatment as assessed by estimated glomerular filtration rate (eGFR) in kidney transplant recipients with caAMR.
DesignThe INTERCEPT study is a phase III investigator-driven randomized controlled open-label parallel arm multicenter study that will examine the efficacy of add-on treatment with TCZ to our SOC in comparison with the SOC alone in the treatment of caAMR. The open-label design was chosen due to the simpler logistics as well as the prohibitive expense of creating a placebo injection, especially since it is an investigator-initiated trial.
Study settingThis in-progress investigator-initiated trial is including transplant recipients who underwent transplantation in Sweden or are currently living here and being followed up at any of the Swedish transplant centers or their affiliated regional centers. The Transplantation Center at Sahlgrenska University Hospital, Vastra Gotaland Region, is the sponsor of the study (contact: niclas.kvarnstrom@vgregion.se). The study sites include three major transplant centers at university hospitals in Gothenburg, Stockholm and Uppsala. Study drug TCZ will be prescribed by the participating sites.
Eligibility criteriaIn the study, 50 adult kidney transplant recipients who have a biopsy-proven diagnosis of caAMR according to Banff 2019 criteria [5] and are at least 12 months after transplantation will be included.
The inclusion and exclusion criteria are listed in Table 1. All biopsies with a diagnosis of caAMR performed for clinical indication will be reviewed at the main study center by two pathologists, and the patients meeting the inclusion criteria and without any exclusion criteria will be considered for the study. Repeat biopsy needs to be performed at randomization if the last biopsy is older than 12 months (+2 weeks) at randomization. In potential patients treated for caAMR with interventions like IVIG, plasma exchange, rituximab, T-cell depleting agent and/or any other medication (including another investigational drug) after the historical biopsy, a protocol transplant biopsy will be performed 2 months ± 2 weeks after treatment completion to confirm the diagnosis of ongoing caAMR. Only patients with eGFR ≥ 20 mL/min/1.73 m2 will be included since a greater impairment of the kidney function may mean an irreversible kidney damage. The screening eGFR determining inclusion should not be older than 1 month at the time of randomization. Infections are a concern of increased overall immunosuppression with the addition of TCZ to the SOC treatment in transplant recipients, which means that patients treated with TCZ may be at an increased risk of infections. Reactivation of viral and other serious infections (e.g. Ebstein–Barr virus (EBV) or tuberculosis (TB)) has been observed with TCZ. Therefore, to avoid the increased risk of infections, patients who are EBV-negative or with a history of TB, active/latent TB or have any active bacterial/viral infection or a history of recurring infections requiring hospitalization in the past will be excluded from the study. At screening, patients with cytomegalovirus (CMV) viremia (without any active disease) may be treated and should have at least two negative CMV-PCR values 1 week apart before they can be randomized. Similarly, patients with low-level BK-virus (BKV) viremia (≤103 copies/mL or < log103) at screening should have at least two negative BKV-PCR values 1 week apart before they can be randomized.
Table 1 Inclusion and exclusion criteriaMoreover, due to already known side effect of TCZ such as leukopenia, thrombocytopenia and hepatic dysfunction, patients who exhibit abnormal liver function tests (alanine transaminase (ALT), aspartate transaminase (AST), bilirubin > 1.5 × upper limit of normal), other significant liver disease as per investigator’s opinion, neutropenia (<2 × 109/L) or thrombocytopenia (<100 × 109/L) will be excluded. Woman of childbearing potential who is unwilling/unable to use an adequate and safe method, e.g. hormonal therapy, to avoid pregnancy for the entire study period and for up to 8 weeks after the last dose of study drug will also be excluded.
ScreeningScreening will take place within 6 months before the randomization. All relevant information will be obtained from their electronic medical charts as well as databases of the respective transplantation centers, pathology and clinical histocompatibility laboratories. Physical examination, chest X-ray, study-related blood samples (including mGFR using iohexol clearance test) as well as a urine sample will be taken, and each subject’s eligibility will be established by a physician at the respective study center before inclusion and randomization of a patient to treatment arms. In patients with an eGFR between 20 and 25 ml/min/1.73 m2 or experiencing a rapid decline in kidney function, eGFR will be repeated at the discretion of the treating physician. In patients who are deemed ineligible for participation at screening, rescreening will be considered on an individual basis and must first be approved by the sponsor.
RandomizationEligible participants, after they have provided informed consent, will be consecutively randomized at the point of randomization by the study coordinator at each site. This will be achieved using a computer program embedded in the electronic case report form (eCRF), assigning participants to either of the two study arms in a 1:1 ratio and stratified by study site. The trial is non-blinded. If a subject discontinues their study participation, their subject code will not be reused, and the subject will not be allowed to re-enter the study again. Randomization will be to one of two treatment groups as follows: Arm A (SOC + TCZ): SOC (as below) + TCZ (162 mg every week, SC administration). Arm B (SOC): tacrolimus (target concentration 6 ±1 μg/L) + MPA (1–2 g/day as tolerated) + steroids (prednisolone not less than 5 mg/day), all oral administration.
The patients who do not tolerate either tacrolimus and/or MPA in the SOC treatment in either arm may be treated with other immunosuppressive drugs. The following regimen will be considered equivalent:
a)Tacrolimus can be replaced with another calcineurin inhibitor, cyclosporine, where a target concentration of 80–120 ng/ml.
b)MPA can be replaced with another antimetabolite, azathioprine (optimally tolerated dose based on leukocyte count) or an mTOR inhibitor, everolimus, with a target concentration of 3–4 μg/l.
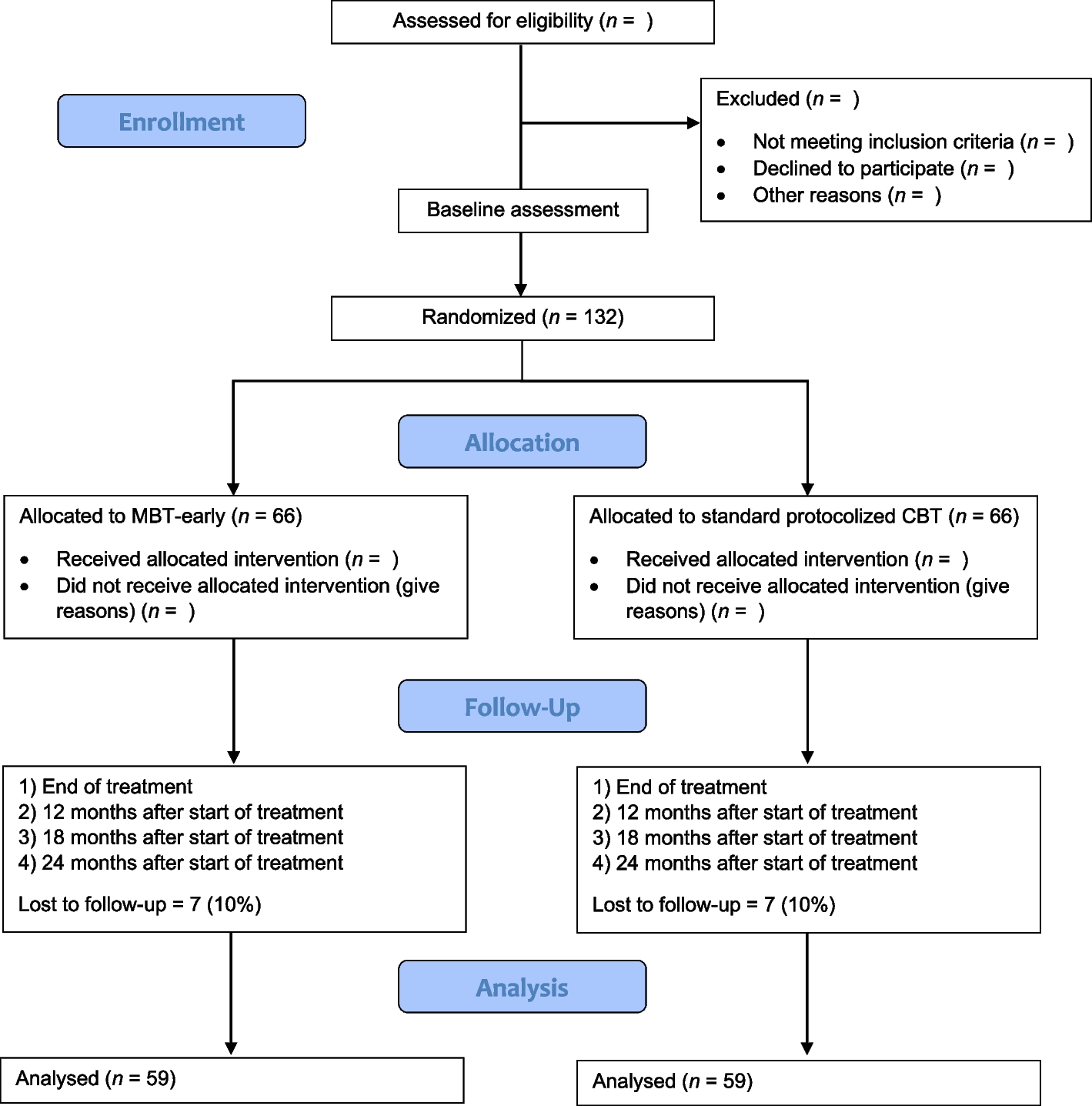
The study treatment will be continued for 24 months after which both groups will continue only with the SOC. All patients will be followed up for an additional 12 months. A study design flowchart is shown in Fig. 1 and a detailed intervention plan can be found in Table 2, the SPIRIT figure. Enrolment will be continued until the required sample size is achieved.
Fig. 1
Flow chart of the Intercept-study. DSA donor-specific antibodies, eGFR estimated glomerular filtration rate, KTx kidney transplant, m months, mGFR measured glomerular filtration rate, MPA mycophenolic acid, sc subcutaneous, SOC standard of care, UACR urine albumin:creatinine ratio
Table 2 The SPIRIT figureSample sizeThe sample size calculation is based on our preliminary analysis of caAMR patients at the SU and assumption of an initial mean eGFR of 48 ± 15 ml/min, with a mean decline (slope) of −7.5 ml/year, a standard deviation of 15 throughout the visits and an intra-patient correlation of 0.85. To uncover a difference of 5 ml/year in the eGFR slope in the two arms, 25 patients would be required in each arm including 10% dropouts, using 1000 simulations, with a power of 80% at a significance level alpha of 0.05, based on eGFR measurements every 3 months using the above specified repeated measures linear model. The sample size is based on decline in eGFR only and therefore, no corrections for multiple comparisons are required.
Feasibility of patient recruitmentThe study’s patient population encompasses all kidney transplant recipients in Sweden, involving all four transplant centers with approximately 6000 active recipients and around 450 kidney transplants conducted annually [28]. Therefore, this sample size is considered achievable due to expected throughput of eligible participants from all centers. In addition, we have implemented various surveillance strategies and new clinical routines for active screening of DSA and performance of early biopsies in transplant patients to facilitate early diagnosis and identification of potential candidates.
InterventionTCZ as a SC regimen will be self-administered by the patients on the same weekday and at the same time. Prefilled injection pens of TCZ will be prescribed electronically to the patients by the study centers. Guidelines for at-home administration of SC injections of TCZ will be implemented in this study in accordance with the routine clinical practice in patients with rheumatoid arthritis. These include training of patients in SC TCZ administration for the first injection by the study coordinators. Adherence to the treatment will be evaluated in congruence with each study visit by patient interviews, assessment of the patient diaries and counting of returned used injection pens.
Close follow-up of all the study subjects will be performed to control or mitigate potential risks. Subjects shall be closely monitored clinically after start of intervention (baseline) at each clinical visit. Physical examinations will be performed, and blood and urine samples will be taken regularly and evaluated. In order to promote participant retention and complete follow-up, the study coordinators will have close contact with the patients via phone and shall send written reminders for every visit and annual protocol biopsies to patients in both arms as well as their regional outpatient clinics. The sponsor will closely monitor recruitment rates and ensure data completeness, through a combination of on-site visits and central monitoring at study sites.
Prohibited/permitted interventionsSince there is no established treatment for caAMR, the patients in the SOC arm will not receive any added treatment. The following treatments are prohibited for the duration of the study in either arm for the treatment of caAMR: any other anti-IL-6/IL-6R monoclonal antibodies, eculizumab, proteasome inhibitors, IVIG (except for treatment of hypogammaglobulinemia or BKV infection during the study) or plasma exchange. For acute T cell-mediated rejection, intravenous methyl prednisolone or T-cell-depleting agents (in case of steroid resistance) may be used for treatment during the study.
Risk mitigationIf a bacterial infection is suspected in any patient, in addition to the routine markers of infections such as white cell counts and CRP, levels of procalcitonin, a marker of infection released by alternate pathways, which should not be affected by IL-6 inhibition, will also be checked in blood [29]. Specific dosage modifications will be made or the drug discontinued in case of adverse events such as cytopenia and elevated liver enzymes.
Furthermore, patients will be made aware of the symptoms potentially indicative of diverticular disease and instructed to alert their healthcare provider as soon as possible if these symptoms arise due to the acknowledged side effect of gastrointestinal perforation. Should this develop, TCZ will be discontinued. Risk factors for cardiovascular disease (e.g. hypertension, hyperlipidemia) will be managed as part of their routine care. Due to interactions of TCZ with statins and calcium channel blockers, patients on these drugs might need dose adjustments. Dose modifications will also be done for neutropenia, thrombocytopenia, elevation of liver enzymes or hypofibrinogenemia which are common side-effects of TCZ. If eGFR falls below 20 ml/min, the decision to continue tocilizumab (TCZ) should be at investigator’s discretion, focusing on participant safety. Significant eGFR reduction may prompt a biopsy for identifying graft dysfunction causes. The study drug treatment will be discontinued prematurely if the patients experience any of the following: pregnancy, diverticulitis, gastrointestinal perforations, persistent neutropenia < 0.5 cells × 109/L, thrombocytopenia < 50 cells × 109/L, elevated liver enzymes ALT or AST > 3 to 5 × ULN or low fibrinogen levels < 50% of the lower limits of normal or malignancies (except local BCC or SCC of the skin or carcinoma in situ of the cervix uteri that have been excised and cured). Study drug treatment may also be discontinued prematurely at the investigator’s discretion due to, e.g. other adverse events suspected to be related to TCZ or protocol violation. Clear guidelines for dose modifications or discontinuation of TCZ are provided in the study protocol. Patients discontinuing study treatment prematurely will continue participating in the study, attending visits and following protocols. They will be allocated in the intent-to-treat (ITT) group. Study termination criteria include loss to follow-up (e.g. due to relocation) or complete withdrawal of consent for further data collection from medical charts.
Outcome parametersPrimary endpointThe primary endpoint is the change in slope of estimated glomerular filtration rate (eGFR) at 24 months after start of treatment using MDRD formula [30] without adjusting for the race component in kidney transplant recipients with caAMR.
Secondary endpointsThe secondary endpoints are specified in Table 3.
iBOXChange from baseline in the mean composite iBox score will be determined at 12 and 24 months after start of treatment. The full iBox risk score is based on the following eight clinical/histological/immunological risk factors: time from transplant to evaluation, eGFR, proteinuria, four histological parameters based on Banff scoring (interstitial fibrosis/tubular atrophy, microcirculation inflammation, interstitial inflammation and tubulitis, transplant glomerulopathy) and iDSA MFI category (Table 4) [20]. The iBox score will be generated using a computerized algorithm and the mean score will be assessed as a continuous variable. An improvement/stabilization of score will be considered as response to treatment and will also be assessed as a yes/no categorical variable. A simplified iBox score based only on four functional and immunological data will also be assessed.
Evolution of DSAThe testing for DSA will be performed in the tissue typing lab at the SU using whole blood samples. Commercially available single antigen flow-bead (SAB) testing One Lambda, Canoga Park, CA (USA), will be used to test for donor specificity, mean fluorescence intensity (MFI), HLA-class and complement C1q-binding capability. DSA MFI is measured as a continuous variable and considered positive if MFI is > 1000. If several DSAs are present, the cumulative MFI (cMFI) will be evaluated by adding together the MFI of each single antibody, as well as the MFI of immunodominant DSA (iDSA), which are defined as the strongest DSAs (the ones with highest MFI) detected in the patients’ sera. Response will be defined as reduction/stabilization in cMFI and/or iDSA at 24 months compared to baseline and assessed as a categorical variable.
Protocol transplant biopsiesAfter the inclusion biopsy at baseline, ultrasound-guided percutaneous protocol biopsies will be performed at 12 and 24 months, after exclusion of a coagulation disorder or thrombocyte count below 80% of the normal value. Three cores of biopsies will be taken with a 16 gauge. All biopsies will be sent to the respective pathology laboratories for further processing and evaluation. The pathologists are blinded regarding which arm the patients are in. Biopsy evaluation will be made through standard paraffin-embedded sections including immunohistochemical complement C4d staining, using a monoclonal antibody (BioSite, rabbit monoclonal antibody, clone A24-T). A diagnosis of caAMR will be made if there is evidence of being both chronic and active, presumably antibody-mediated tissue damage (Table 5), as defined by the Banff 2019 criteria [5] and the biopsies will be scored accordingly.
Table 5 Diagnostic criteria of caAMRaAll biopsies which show aAMR without evidence of chronic changes by light microscopy will be assessed by electron microscopy in order to exclude or confirm presence of peritubular capillary basement membrane multilayering or glomerular basement membrane double contours. If the criteria for caAMR are no longer fulfilled in the follow-up biopsies, response to therapy is assumed. The response will be assessed as a yes/no categorical variable. In all biopsies, which still meet the required criteria for caAMR, means of individual Banff lesion scores will be compared between the baseline biopsy and the 12- and 24-month biopsies as continuous variables.
QuestionnairesWe also aim to assess in this study the three PRO mentioned above. We will also evaluate the differences in these PRO in relation to age, sex, occupation, civil status, educational status and type of treatment arm, and fear of graft rejection in relation to biopsy or type of treatment arm at baseline and at 12, 24 and 36 months. The following validated qualitative research instruments will be self-reported by the patients, either directly online or on paper, at baseline and every 12 months until 36 months. Permissions to use these instruments in this study have been obtained.
a)The OTSWI (Organ Transplant Symptoms and Well-being Instrument)
The OTSWI is a multi-item questionnaire that has been developed to measure symptom prevalence, symptom distress and transplant-specific well-being after organ transplantation. It will be used to assess transplant-specific symptoms and well-being during the whole study period [24].
b)The BAASIS© (Basel Assessment of Adherence with Immunosuppressive medication Scales) is a self-report instrument for assessingherence to immunosuppressive drugs. Possible associations between non-adherence and rejection and/or development of DSA will be explored [26].
c)The PTGR (Perceived Threat of the risk of Graft Rejection) is a multi-item questionnaire that measures the phenomenon labelled, ‘graft-related threat’ (GRT), ‘intrusive anxiety’ (IA) and ‘lack of control’ (LOC). Possible associations between the three different PTGR parts and OTSWI and rejection will be explored [25].
Statistical analysisThe primary analysis of eGFR decline by therapy will be performed using a repeated measures linear model (two sided at alpha 5%) adjusting for eGFR at baseline and donor status (living versus deceased) as fixed effects and center as random effect. Visits/time will be included in the model as a linear variable and the P-value of the interaction term of decline in eGFR and visit/time will be the primary analysis. The estimated treatment difference with the associated 95% confidence interval (CI) and P-value will be presented. Two-sided P-value less than 0.05 will be considered significant.
The hierarchical testing procedure below is introduced to guarantee that the probability of Type 1 error is <5% for all confirmative statements. The order of the hierarchical testing procedure will be as follows:
1.eGFR decline (primary efficacy analysis)
2.Change from baseline in mean composite iBox risk prediction score at 24 months
If the first analysis is significant, the probability mass 0.05 will go to the second analysis. If the first analysis is non-significant, no analysis will be confirmative.
Results from the other endpoints and from subgroups shall be considered hypothesis generating only. P-values for the endpoints other than eGFR (primary) and iBox, if presented, should be interpreted in a descriptive fashion only and cannot be considered as significant.
Secondary endpoints will be analysed in an exploratory manner, using appropriate parametric and non-parametric statistical methods. For comparisons between two groups Fisher’s exact test will be used for binary response data, Mantel–Haenszel chi square test for ordered categorical data, the t-test for independent samples or Mann–Whitney U test for continuous data. Continuous data will be expressed using mean (standard deviation) or median (interquartile range) and categorical data as numbers (frequencies); 95% confidence intervals will be calculated when appropriate. Missing data will be analysed with regard to reasons and pattern, and sensitivity analyses will be performed based on various assumptions regarding the pattern. The results from the primary and secondary endpoints will also be stratified and presented according to sex.
The ITT population will consist of all randomized patients who take at least one dose of assigned treatment and have at least one follow-up with measurements. The analysis of all efficacy data will be performed on the ITT population. The per-protocol (PP) population will include all ITT patients without any major protocol deviations, who did not have to stop the study drug for >4 weeks due to side-effects, and who had ≥80% compliance with study drug (with dose adjustments if required) while on treatment (up to discontinuation for patients whose treatment is terminated early). The PP analysis will be used to assess the robustness of the ITT analysis results of primary and secondary efficacy data.
The statistical analysis will be performed using the commercially available software SAS v9.4. A detailed statistical analysis plan will be written where all populations, variables, and statistical methods will be described. The total number of subjects per arm (n = 25) considers a total dropout rate of approximately 10%. These patients will still be included in the final ITT analysis. For the patients who drop out, their data will be analysed up until the date of last available clinical data.
Data Monitoring and Safety BoardTo secure the safety of the INTERCEPT study population and integrity of the study, the totality of data will be reviewed on a regular basis by an independent Data Monitoring and Safety Board (DMSB). The DMSB will consist of two physicians and one statistician, neither with any other involvement in the study. The first formal interim analysis meeting will be held to review data relating to treatment efficacy, patient safety and quality of trial conduct when approximately 40% of the patients (~20 subjects) have reached the time-point for the primary endpoint assessment and the data are cleaned. Subsequently, meetings will be held after the inclusion of all patients is complete, upon the attainment of the primary endpoint in all enrolled patients and in the event of any safety concerns. The DMSB will determine if amendments to the protocol or changes in study conduct are required and may consider terminating the trial if there are major safety concerns.
Quality control and assuranceThe study will undergo thorough monitoring by an independent study monitor before, during and after the study to ensure protocol adherence and proper collection, documentation and reporting of that data and all essential documents. This will be conducted in accordance with International Council for Harmonisation (ICH)-GCP E6 (R2) standards and applicable ethical and regulatory requirements. The investigator will provide access to all source documents, including eCRFs and other protocol-related materials. Patient confidentiality will be maintained per local regulations. The monitor will regularly review eCRFs based on a defined monitoring plan to verify adherence to and completeness of protocol as well as the validity and accuracy of entered data. Throughout the study, the monitor will conduct predefined visits at the study center, verifying informed consent, adherence to inclusion/exclusion criteria, documentation of severe adverse events and the recording of the main efficacy, safety and tolerability endpoints.
Data managementAll data collection in the study will be through an eCRF. Investigators are responsible for accurate data registration and corrections per the study protocol. The independent external monitor will randomly verify data accuracy by comparing it with source documents. This verification ensures compliance with the International Council for Harmonisation (ICH)-GCP E6 (R2) and relevant guidelines and ethical regulations. If no inconsistencies are found, the appropriate eCRFs are collected. The investigator will sign the completed eCRF, and a copy of it will be archived at the study sites. Queries or responses will be processed into the database by the Data Management team.
Investigators are responsible for ensuring that all data in the eCRFs and Data Clarification Forms are accurate, complete and legible, and that all entries are verifiable with source documents. Source documents for each subject in the study will be retained, and a document defining classified source data will be included in the Investigator Site File (ISF) at each site. Data management will be overseen by the sponsor representative/PI. The study database will be soft-locked upon receipt and cleaning of all specified data in the study protocol. It will be hard-locked after a (blind) data review meeting, where all data-related decisions have been made and reflected in the database.
Ethical considerationsThe study will be executed in alignment with the study protocol, ICH-GCP E6 (R2), the latest DMSB ensure the safety and integrity of the study subjects as well as the quality of the data collected. Patients who are willing to participate will be given adequate oral and written information about the study, its purpose, any risks and benefits as well as inclusion and exclusion criteria, as well as the informed consent form (ICF) by licensed physicians at the study center. In case any new ancillary studies are planned with the already stored biological samples, a new informed consent will be obtained. Each subject who participates in the study will be identified by a subject number on a subject identification list. Any amendments that occur throughout the study will be passed on to affected parties.
Patient data and samples for this study will be handled with confidentiality measures and GDPR compliance. Each study participant will be assigned a subject number for identification, and blood and urine samples collected for future biomarker research will be coded with a unique study identification number. The identification/code list will be securely stored in locked cupboards at the Clinical Trial Units of the respective Transplantation Centers to prevent unauthorized access. Only study personnel will have access to the code list. Samples collected from study patients will adhere to the Biobank Act and be registered with Biobank West (www.biobankvast.se), under Region Vastra Gotaland. The samples will be assigned a unique QR code in the biobank, linked electronically to personal numbers and stored pseudonymized to protect participant identification. Access to the code key and samples will be restricted to study personnel only. Samples will be stored for at least 10 years post-study, used for study-related purposes, and then destroyed. Identification lists will be kept securely for the same duration and destroyed afterward.
All data, including informed consent, completed eCRF, protocol and final report, will be encrypted for accurate reporting and stored at the study center for at least 10 years post-trial, as per Swedish law. Information processed by the sponsor will be pseudonymized with a study identification number, ensuring anonymity in the presentation or publication of study results.
The investigators intend to communicate the trial results to participants, healthcare professionals and the public via a summarized manuscript published in a scientific journal. All subjects are insured through the Swedish patient insurance and will receive post-trial care if they suffer any harm from trial participation. Paid sick leave is possible if deemed necessary.
Study registrationThe final study protocol including the final versions of the informed consent form and other information provided to subjects for the INTERCEPT study have been approved by The Swedish Ethical Review Authority (Etikprövningsmyndigheten, Dnr 2020-03156). It has also been approved by the Medical Products Agency (Läkemedelsverket, Dnr 2019-004302-10). The study has been registered in a public clinical trial database, ClincalTrials.gov (NCT04561986).
留言 (0)