
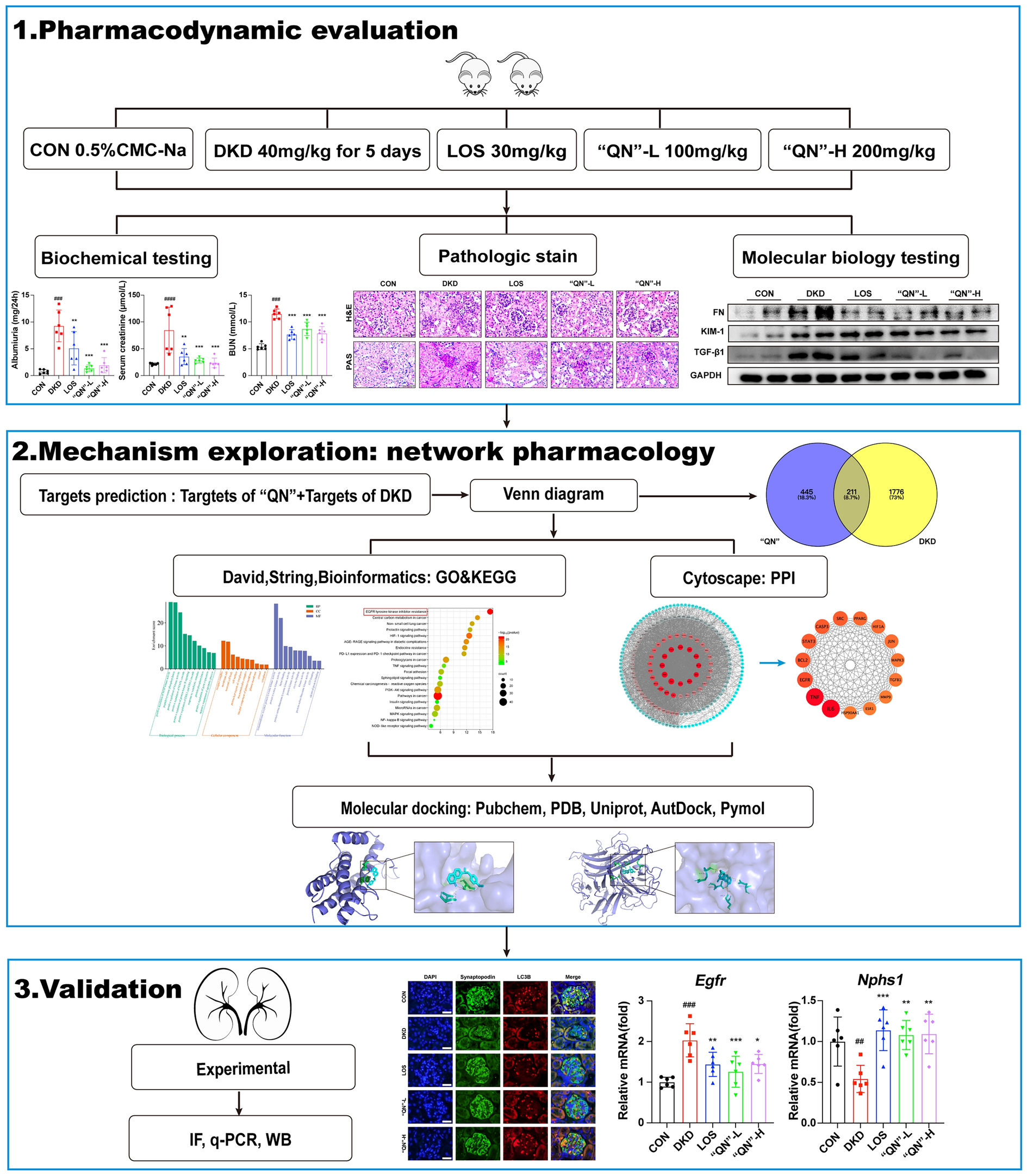
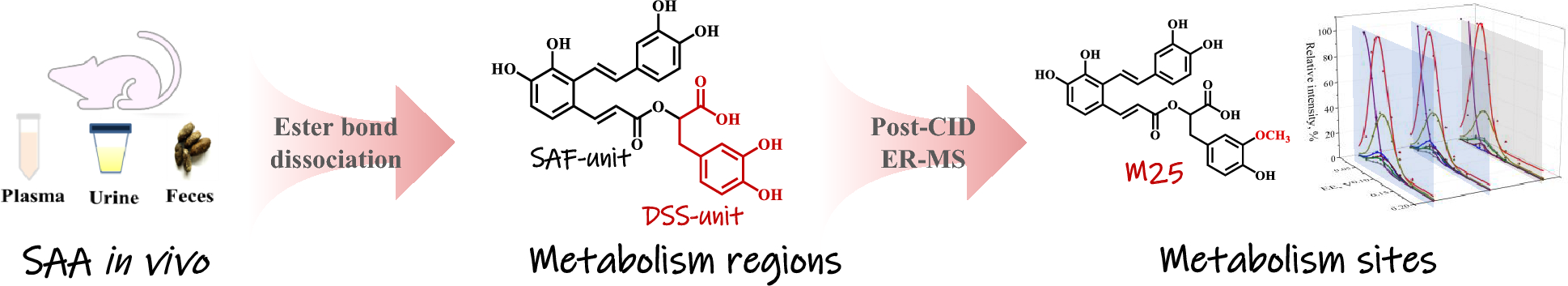
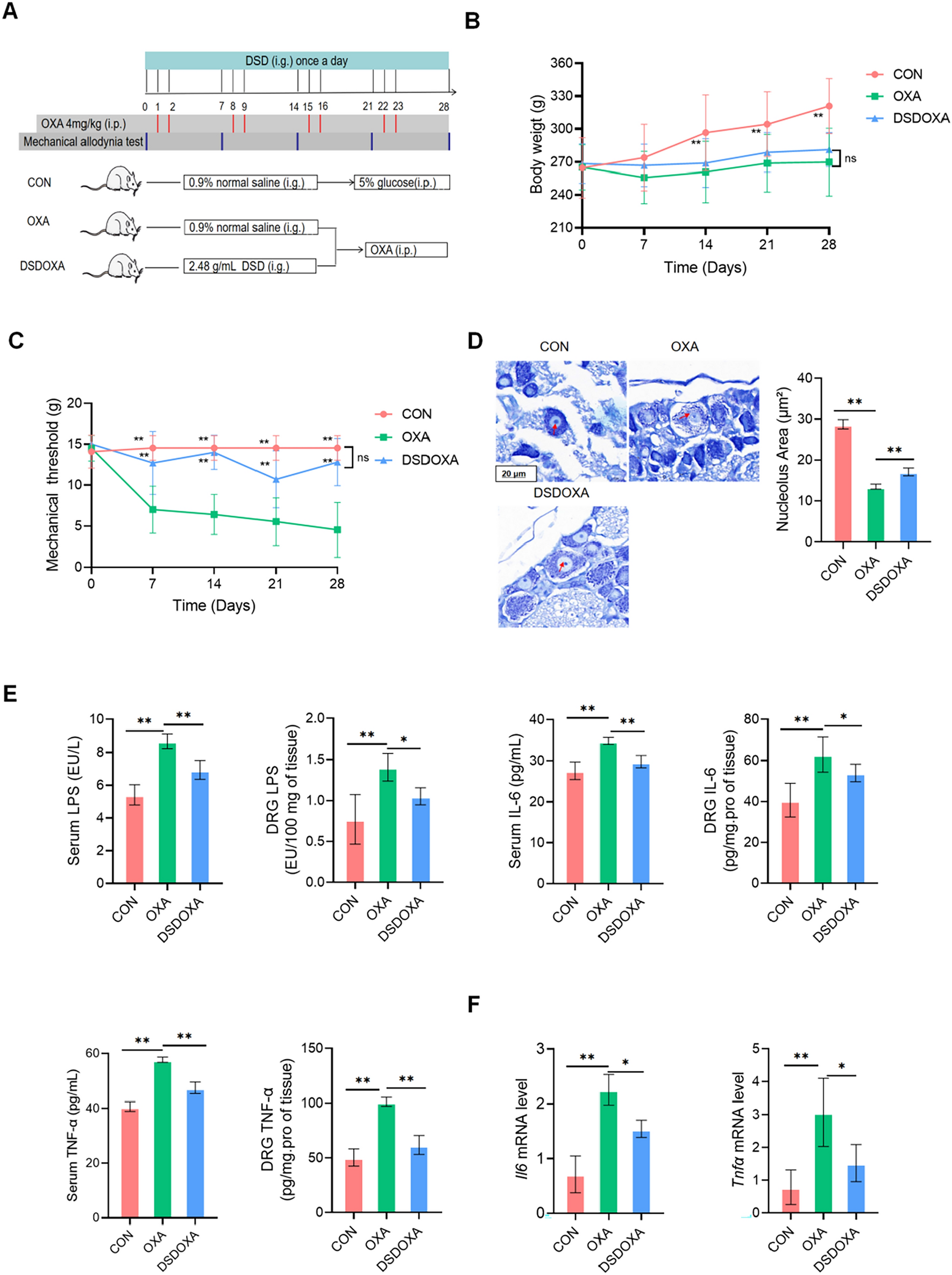
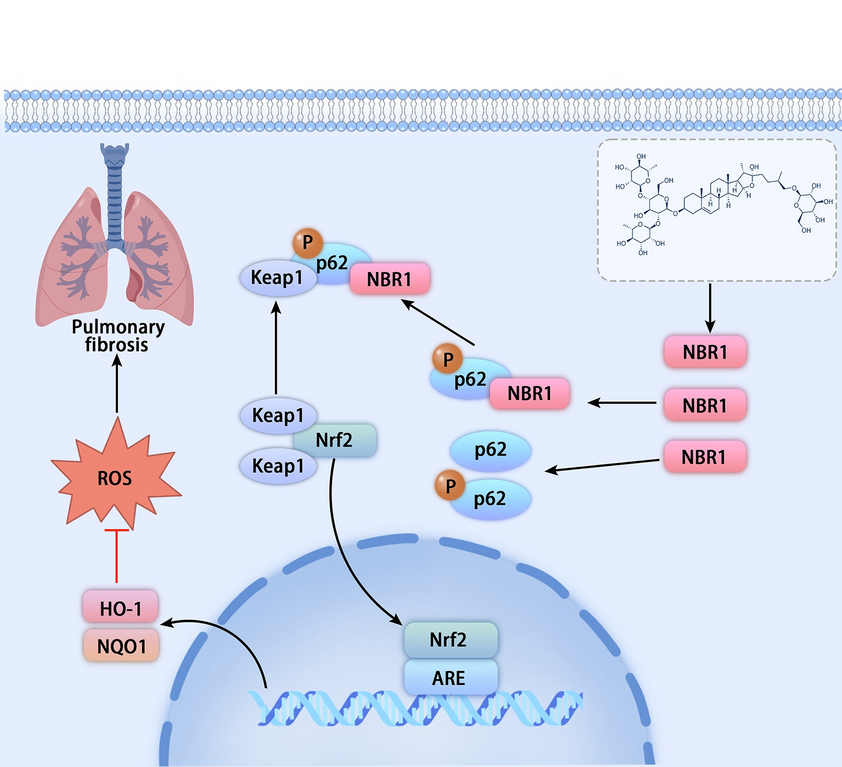
記住我



TCMNPAS is a web-based platform developed using the Shiny framework and HTML. It aims to facilitate comprehensive analysis and research in the field of TCM.
TCMNPAS offers a range of powerful tools and functions, primarily organized into eight main function panels, as depicted above (Fig. 3).
Fig. 3
Databases included in TCMNPAS v1.0 for searching herbs, components and targets (A), Setting the threshold of QED value for drug-likeness and setting the threshold of drug-target score (B), Setting the significance P-value of drug-target (C), Herb-Compound-Target (D), Setting the violation count of Lipinski Rule and Veber Rule for drug-likeness (E), KEGG Enrichment (F), Shared-KEGG-Enrichment-Curve (G), Network Distance Score (H), KATZ Score (I)
Formula mechanismThe analysis of the chemical characteristics of herbal formulas in TCMNPAS is achieved by inputting a list of herbs, which can be specified using their Chinese name, Pinyin name, English name, or Latin name (Fig. 3A).
In the quest to identify bioactive ingredients within herbal formulas, TCMNPAS employs the quantitative estimate of drug-likeness (QED) presented by Bickerton [41] as a key metric for drug-likeness screening. QED integrates eight essential molecular descriptors for effective analysis of drug-likeness in pharmaceutically active compounds within the formulas [20,21,22, 41,42,43], as depicted in Fig. 3B. Additionally, the Lipinski rule [44] and Veber rule [45] are also incorporated for drug-likeness screening. The Lipinski rule assigns values from 0 to 4, representing the number of violations against the rule, with a higher number indicating poorer drug-likeness. According to the “Rule of Five”, a drug-like molecule should have no more than one of the following violations: (1) No more than 5 hydrogen bond donors; (2) No more than 10 hydrogen bond acceptors; (3) Molecular weight no more than 500; (4) LogP no more than 5. Similarly, the Veber rule values range from 0 to 2, indicating violations against the rule, with a higher number signifying reduced drug-likeness. According to the “Rule of Veber”, a drug-like molecule should have no more than one of the following violations: (1) No more than 10 rotatable bonds; (2) Polar surface area of no more than 140 or no more than 12 hydrogen bond donors and acceptors (Fig. 3E).
In TCMNPAS, the identification of core targets for herbal formulas is a crucial step achieved through target profiling of formula ingredients. The determination of core targets relies on a threshold score for a compound-target association, primarily based on the scores obtained from the STITCH database. A higher score in STITCH indicates a stronger association, with a median score of 400. In cases where compound-target pairs lack association scores in other databases, a uniform value of 9999 is assigned [22, 33].
TCMNPAS utilizes a binomial statistical model to facilitate the assessment of target profiling for formulas. This model calculates the probability P(X ≥ k) of a target interacting with k or more active compounds. A target with a smaller P value (e.g., P < 0.05) indicates a significantly larger observed number of interacting compounds, suggesting its role as a core target for the formula [20,21,22]. The score for a specific target of herbal formula (geneScore) is calcucated by using a numerator that equals the negative logarithm of P(X ≥ k) and a denominator that equals the rank of P(X ≥ k) as follows [20,21,22]:
$$geneScore = \left\ \frac},\,if\;P(X \ge k) < P_ \hfill \\ 0\;,\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;otherwise \hfill \\ \end \right.\;\;\;\;$$
(1)
The threshold for identifying core targets of a herbal formula, denoted as Psig, is a user-defined value. This threshold plays a crucial role in the identification of core targets of the herbal formula. Furthermore, the score of a compound, referred to as chemScore, can be determined by averaging its corresponding target scores as follows [20,21,22]:
$$chemScore = \frac }}\sum\limits_^ }} } \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;$$
(2)
TCMNPAS empowers researchers to characterize the functional profile of formula targets through enrichment analysis. This analysis includes Gene Ontology (GO), KEGG pathways, Reactome pathways, and Disease Ontology (DO). The hypergeometric distribution model is utilized for the enrichment analysis [35, 40]. To ensure statistical significance, the False Discovery Rate (FDR) method is employed to adjust the P values. The enrichment analysis is conducted using the “clusterProfiler package” and “DOSE package” based on R software [20,21,22, 40, 46] (Fig. 3C).
Additionally, TCMNPAS facilitates the analysis of the association between diseases and formula targets. By providing disease targets, the platform performs a co-association analysis of the enriched terms between formula targets and disease targets. The “Shared-GO-Enrichment-Curve,” “Shared-KEGG-Enrichment-Curve,” and “Shared-Reactome-Enrichment-Curve” options display co-association curves, allowing users to adjust the number of co-associated terms. TCMNPAS provides co-association scores and AUC values for shared term curves, aiding in the evaluation of the degree of association between formula targets and disease targets. This information enables researchers to infer the potential roles of formula targets in disease treatment (Fig. 3D, F-G).
Targets mechanismResearchers utilizing the system have the flexibility to customize the name of the target group and input standard Entrez GeneIDs directly or use text files containing GeneIDs for target group analysis. Furthermore, they have the option to provide text files containing disease-related targets (".txt" or ".csv") with one ID per line, using standard Entrez GeneIDs. When disease targets are inputted, the analysis results will showcase the corresponding molecular mechanisms of the disease targets, allowing for comparison with the inputted target group and highlighting them as “Disease” in the results (Additional file 1: Figure S1).
Upon inputting disease targets, the system presents tabs for “Shared-GO-Enrichment-Curve” and “Shared-KEGG-Enrichment-Curve”, which display co-enrichment curves. Additionally, the “GO-MF-Enrichment,” “GO-BP-Enrichment,” “GO-CC-Enrichment,” “KEGG-Enrichment,” “Reactome-Enrichment,” and “DO-Enrichment” tabs concurrently exhibit co-enriched scatter plots of both the formula and the disease. The co-enrichment terms are displayed in this section for comprehensive analysis.
“Formula Targets”, “Formula Compounds”, “Herb-Compound-Target”, “Shared-GO-Enrichment-Curve”, “Reactome Enrichment”, “Shared-Reactome-Enrichment-Curve” and “DO Enrichment” results are shown in Additional file 1: Figures S2-8.
Network associationIn the context of TCMNPAS, relevance inference relies on two critical scores: the KATZ score [26] and the network distance score [21, 22]. These scores are applied to assess the relevance between formula targets and disease targets based on their connectivity within the Protein–Protein Interaction (PPI) network integrated by TCMNPAS, derived from the HIPPIE (Human Integrated Protein–Protein Interaction Reference) database [34].
The KATZ score is a relevance score that considers the distance and path between network nodes, with a path score coefficient (Beta) of 0.001. The output of the KATZ score includes various specific score items such as overlap score, Path 1 score, Path 2 score, Path 3 score, Total Score, Random Score Medium, and P-value (Fig. 3H). On the other hand, the network distance score represents the shortest path length in the PPI network. The specific items included in the network distance score output are mean distance, mean random score, and P-value (Fig. 3I).
Both the KATZ score and the network distance score serve as essential metrics for evaluating the relevance between formula targets and disease targets, with higher relevance scores indicating closer proximity between them in the PPI network.
Formula compoundsTCMNPAS offers a comprehensive formula compounds retrieval module, encompassing 1630 commonly used herbs and 18,090 compounds (including their chemical structures) [21, 22, 42, 43]. Users can retrieve formula compounds by inputting herb lists with Chinese names, Pinyin names, English names, or Latin names. Additionally, TCMNPAS allows the retrieval of compound information by entering the chemical structure representation of a compound (InChIKey, e.g., ZYGHJZDHTFUPRJ-UHFFFAOYSA-N). This function facilitates the retrieval of information about the compound in various herbs (Additional file 1: Figure S9) [15, 16, 22, 33]. Researchers can further explore the retrieved compounds for their properties or structures by utilizing additional sources such as PubChem [47].
Network visualizationIn TCMNPAS, researchers can easily visualize the Herb-Compound-Target network. The required network file is available in CSV format, can be downloadable from the “Herb-Compound-Target” tab in the Formula Mechanism module.
The integrated PPI network in TCMNPAS is derived from HIPPIE. By selecting the “Seed-expansion in PPI network” option, corresponding targets are projected onto HIPPIE, leading to the formation of subnetwork outputs [21, 22, 34]. Additionally, the system offers three network types to choose from, with the option to enable PPI network projection and dynamic display (Additional file 1: Figure S10).
Prescription miningIn recent years, a prescription mining framework based on herb-herb networks has been developed. This framework involves core herbs (combined with network centrality analysis), core herb pairs (combined with the entropy approach), core formulas (combined with the BK algorithm [23,24,25]), and core effective formulas (combined with the GA algorithm and regression model) (Fig. 4A). Notably, several medication rules/guidelines of renowned TCM experts, such as Professor Liu Jiaxiang (Chinese medical master, an expert in oncology) [48, 49], Professor Tang Hanjun (a mammography expert) [50, 51], Professor Xu Rongjuan (an expert in endocrinology) [52], and Professor Chen Yipin (a nephrology expert) [53, 54], have been effectively summarized.
Fig. 4
Prescription mining algorithm (A), parameter setting area for prescription mining analysis (B), Summary of results (C), Core formulas (D), Herb compatibility network (under optimized threshold) (E), Optimized herb compatibility network (F)
To expedite the rapid analysis of core herbs, core herb pairs, and core formulas, the platform provides a prescription mining functionality that mainly utilizes the BK algorithm to find the core formulas in herb-herb networks. The basic principle is to find all the maximum cliques based on the recursive procedure for optimizing the candidate-selected herb. The algorithm continuously replaces the herb to continue the search until all herbs have been traversed, thereby obtaining all the maximum cliques in the network. These maximum cliques in the herb-herb network can be considered core formulas.
Individualized treatment in TCM involves formulating therapeutic prescriptions by adding or reducing herbs based on core formulas after syndrome differentiation. Generally, herb-herb networks are weighted undirected networks, where the frequency of herb combinations is used as the edge weight of the network. However, the BK algorithm is only applicable to unweighted networks. Therefore, TCMNPAS performs adaptive binarization on weighted networks before running the BK algorithm and evaluates core formulas based on the two metrics of prescription support and confidence: (1) average confidence of a core formula (α); (2) support under a confidence α, Sα. These two metrics are described in detail elsewhere [55].
In the context of prescription mining analysis using TCMNPAS, researchers need to follow specific steps and set various options for optimal results. The initial step involves uploading prescription data, followed by configuring the S0.9 threshold, the minimum number of herbs in the core formula, and the desired number of herbs. This module has certain formatting requirements for the prescription data uploaded by users: (1) The file format should be CSV in UTF-8 encoding; (2) Prescription data should consist of 3 columns [Patient ID (Pid), Visit ID (Vid), and Herb composition]. Vid is used to identify different visit times. If there is no visit ID, please use the same value. Pid is used to identify different patients. Additionally, users can choose from several options, including “Enforce the core formula containing drug number to be equal to the expected drug number during adaptive screening” and “Merge core formulas with high similarity.” The next step is to determine the method for merging highly similar core formulas, set the similarity threshold for merging core formulas, and choosing options such as “Calculate person-based statistics,” “Visualization of compatibility network,” and “Dynamic display” [25, 42, 55, 56](Fig. 4B).
Firstly, users must set the threshold for S0.9 support, typically between 0.01 and 0.3. A higher S0.9 value indicates higher support for the discovered core formulas; however, it yields fewer amounts. While it's crucial to set the minimum number of herbs in the core formula, one must be careful not to set this number too high, as it could prevent finding core formulas that satisfy the requirements. The binarization threshold also requires optimization during core formula mining. Secondly, users should predefine the desired number of herbs in the core formula, being mindful not to set it too high, as it may result in no core formulas that meet the requirements. Further options include selecting whether the number of herbs in the core formula should be equal to the desired number (maximizing the number of core formulas with the desired herb count) and whether highly similar core formulas should be forcibly merged.
TCMNPAS provides two methods for consolidating highly similar core formulas: “Together,” which merges highly similar core formulas collectively, and “Step,” which incrementally merges highly similar core formulas. The similarity threshold for merging core formulas should be set between 0 and 1, with a recommendation to choose a value greater than 0.6. If the input prescription data includes Vid, selecting the “Calculate person-based statistics” option will facilitate patient-based core formula statistics [25, 55,56,57].
The input file format must adhere to specific requirements. The prescription data should be in CSV format and include three categories (Pid, Vid, and herb). Pid represents the patient ID, used to identify different patients, while Vid represents the visit ID, used to identify different time points. Herb represents the composition of the prescription. Two considerations are crucial during file preparation: firstly, if there is no visit ID, it should be marked with the same value; secondly, attention should be paid to the standardization of herb names in the prescription composition.
This module provides four essential analysis results, namely “Summary of results”, “Core formulas”, “Herb compatibility network”, and “Optimized herb compatibility network” (Fig. 4C-F).
Molecular dockingMolecular docking is a critical bioinformatics technique, that plays a significant role in understanding the interaction between molecules (Fig. 5A). This computational method allows researchers to investigate the binding affinity and spatial arrangement of molecules, shedding light on their potential interactions and functional implications. In the TCMNPAS platform, both single-molecule docking and batch docking modes are provided (Fig. 5B). The platform incorporates the Autodock Vina molecular docking module (open-source software, https://vina.scripps.edu/) [27, 28], which supports Vina and PSOVina [29, 30]. The PSOVina algorithm, an optimized version of Autodock Vina, utilizes a hybrid particle swarm optimization algorithm, achieving higher accuracy and speed compared to Autodock Vina [29,30,31].
Fig. 5
Molecular docking principle diagram (A), Parameter setting and result example page for single molecule docking (B), Cross batch docking mode (C), Parallel batch docking mode (D), Molecular docking results (E), Extraction of standard ligands from PDB (F), Batch retrieval of PDB docking parameters (G), RMSD calculation (H)
To perform molecular docking in TCMNPAS, compounds can be inputted in SMILES or INCHIKEY format. Alternatively, compound files (. mol2 or.pdb) can be uploaded, and the default Vina ligand preparation program [27]will be utilized. The protein structures of specific targets can be obtained and prepared using the “Batch retrieval of PDB docking programs” in the Molecular Docking module [58].
TCMNPAS offers two built-in methods for obtaining protein docking pocket parameters: Fpocket [59] and ligand-based. When using the ligand-based method, it's necessary to input the protein with its native ligand to extract corresponding pocket parameters from the ligand’s position. The platform also facilitates extraction of native ligands from PDB files. For Fpocket prediction, version 2.0 of the Fpocket program is utilized. Alternatively, users can opt to manually input parameters such as center_x, center_y, center_z, size_x, size_y, and size_z. The platform additionally supports batch retrieval of PDB docking parameters and root mean square displacement (RMSD) calculations (Fig. 5F-H).
In the batch docking mode, users can choose between two options: (1) Cross mode [each ligand docked with each receptor (Fig. 5C)], and (2) Parallel mode [ligands paired with receptors for docking (Fig. 5D)]. In parallel mode, it is essential to ensure that the number of ligands matches the number of receptors. TCMNPAS supports multiple scoring functions to evaluate the affinity and stability of molecular docking, taking into account various factors such as binding energy and steric hindrance to improve the reliability of docking results. After docking is completed, TCMNPAS generates a detailed docking report, including binding modes and scoring values for each molecule with the target protein. Users can visualize and analyze the docking results using the provided tools to gain insights into the binding modes and activity of the molecules. Additionally, the docking protocol can be validated in TCMNPAS by calculating the RMSD of redocked poses of native ligands. The final molecular docking result is presented in Fig. 5E.
As an example of ligand docking in TCMNPAS, quercetin was selected [60]. Quercetin’s “Standard SMILES” was retrieved from the PubChem database, and the “Protein Data Bank (PDB) ID” for Akt (3O96) was obtained from the RCSB PDB database (https://www.rcsb.org/). Subsequently, both pieces of information were input into the molecular docking module of TCMNPAS to analyze the binding affinity of quercetin towards Akt1. The results demonstrated a strong binding affinity of − 8.9 kJ/mol, indicating a favorable interaction between quercetin and Akt1 [61].
ToolsThe TCMNPAS system offers a range of valuable tools to facilitate various analyses and data visualizations.
ID Conversion The “ID Conversion” tool offers batch conversion capabilities, allowing users to convert specified variables between Entrez Gene IDs and Gene Symbols, both from entrez gene ID to gene SYMBOL and vice versa. (Additional file 1: Figure S11).
Gene ID to PDB ID With the “gene ID to PDB ID” tool (Additional file 1: Figure S12), users can input gene IDs, and the system will provide corresponding PDB IDs for further exploration.
Seed in KEGG Pathway The “Seed in KEGG pathway” tool [62] (Additional file 1: Figure S13) allows users to input their desired pathway ID, and the system will display the corresponding KEGG pathway diagram, assisting in pathway analysis.
Heatmap The “Heatmap” tool [63] (Additional file 1: Figure S14) provides customizable options, and offers flexible customization options for personalized heatmap visualization.
Data Visualization The “Data Visualization” tool (Additional file
留言 (0)