
記住我

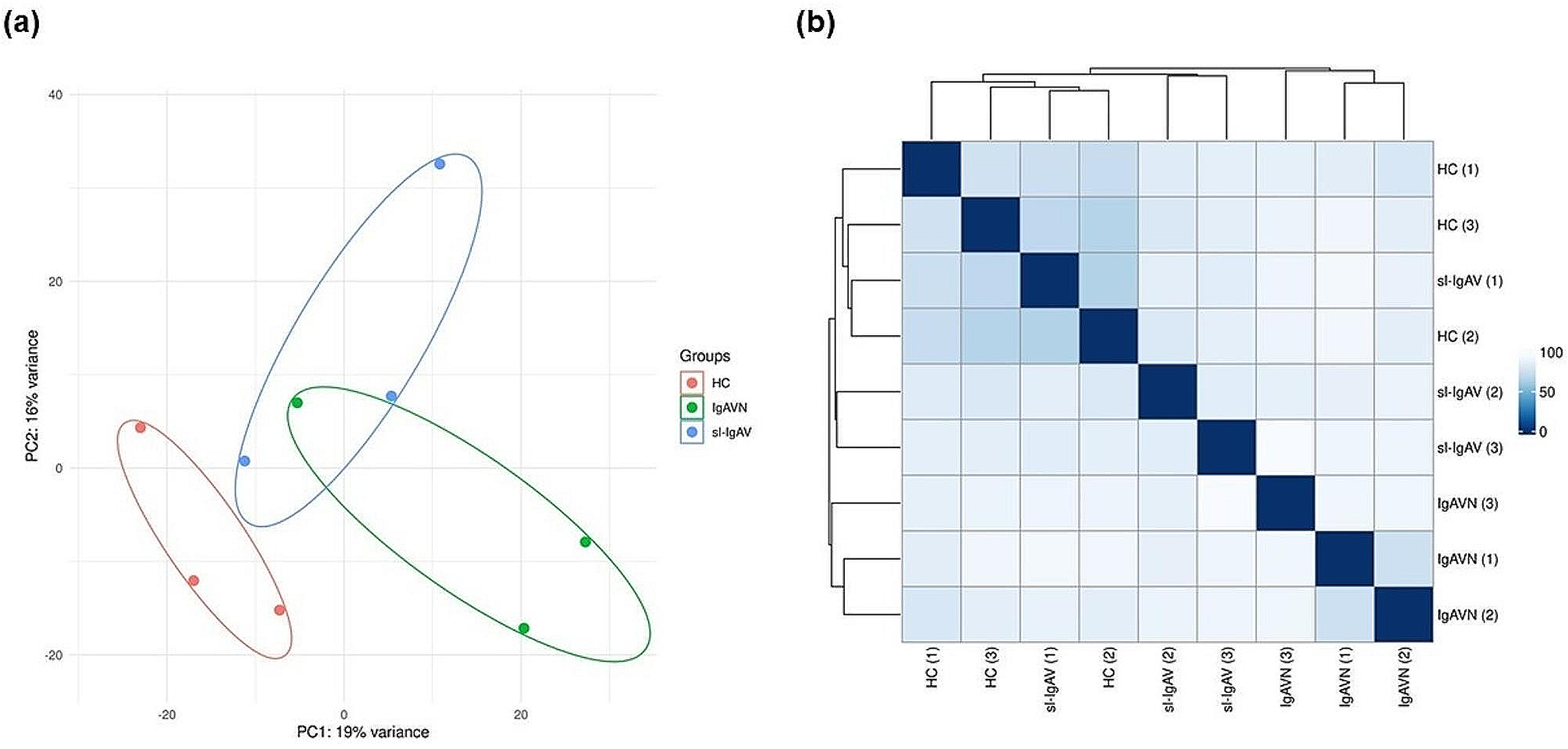
Fourteen SSc patients, followed by the Division of Clinical Rheumatology, University of Genova, Italy, were enrolled following the approval of the Ethical Committee of the Ospedale Policlinico San Martino (237REG2015, amendment number:002–28/05/2018) and the collection of the written informed consent.
The patients were included according to the 2013 American College of Rheumatology (ACR)/European Alliance of Associations for Rheumatism (EULAR) criteria for SSc [23]. Clinimetrics was executed according to good clinical practice guidelines [24]. The presence of ILD was confirmed through a routinely administered high-resolution computed tomography (HRCT) [25, 26].
The definition of ILD in SSc patients has been adapted from the American Thoracic Society guidelines for IPF, which include the radiologic presence of traction bronchiectasis, traction bronchiolectasis, ground-glass opacities, reticulation, honeycombing, other interstitial lung abnormalities, a combination of these findings or any recognized interstitial composite pattern [26]. Based on that, four enrolled SSc patients were not characterized by the presence of ILD, and they were included in the group of “SSc patients no-ILD”. Demographic and clinical characteristics of enrolled SSc patients, including auto-antibodies profile, therapies, and clinical organ involvement are summarized in Table 1.
Table 1 Clinical and demographic characteristics of SSc patients. Clinical and demographic features od enrolled SSc patients affected and no-affected by interstitial lung diseases (SSc-ILD pts and SSc pts no-ILD, respectively). Reported data include age, sex (F/M), disease duration, auto-antibody profile, organ involvement, and therapy. SSc patients without ILD are not displayed in this tableIn particular, the mean age was 63.6 ± 14 years in SSc-ILD patients and 64.7 ± 9.4 years in SSc patients no-ILD; the mean disease duration was 7.2 ± 5.1 years in SSc-ILD patients and 14.75 ± 5.91 years in SSc patients no-ILD; there were 7 female (70%) and 3 male SSc-ILD patients, whereas the SSc patients no-ILD were all female. The most common autoantibody among patients was anti-topoisomerase-I antibody (Scl70). Skin involvement was present in all subjects and was classified according to LeRoy and Medsger criteria [27]: the 50% of patients in both groups had “limited” cutaneous SSc (lcSSc), and the 50% had “diffuse” cutaneous SSc (dcSSc). The mean modified Rodnan skin (mRSS) score was 8.7 ± 6.68 in SSc-ILD patients and 6.11 ± 4.84 in SSc patients no-ILD; all SSc-ILD patients and 3 SSc patients no-ILD had digital ulcers (DUs) or a history of DUs.
Regarding other relevant disease targets in the SSc-ILD group, 4 patients (40%) showed gastrointestinal involvement, one patient (10%) had renal involvement, and no patients had pulmonary arterial hypertension (PAH). In the group of SSc patients no-ILD, all patients showed gastrointestinal involvement, one patient had pulmonary arterial hypertension (PAH), and no patients had renal involvement.
Among the enrolled SSc-ILD patients, the average forced vital capacity (FVC) was 89.7% ± 32.5 of the predicted value, the average forced expiratory volume in 1 s (FEV1) was 87.7% ± 27.5 of the predicted value, and the average diffusing capacity for carbon monoxide (DLCO) was 66.7% ± 28.8 of the predicted value.
All patients were under standard immunosuppressant therapy; in the SSc-ILD patient group, the most used drugs were mycophenolate mofetil (MMF), prednisone (PDN), hydroxychloroquine (HCQ), methotrexate (MTX), and rituximab (RTX). We considered for rituximab not only the current administration of the drug, but also an historical use of it.
From the vasodilatory perspective, 4 patients (40%) were treated with endothelin receptor antagonists (ERA), 1 patient (8.3%) was treated with phosphodiesterase type 5 inhibitors (PDE5-i), and 4 patients (40%) were treated with intravenous prostanoid regimens using iloprost. In the group of SSc patients no-ILD, one patient was treated with HCQ, one patient was treated with iloprost, one patient was treated with ERA, two patients were treated with PDE5i, and two patients were treated with ACEi. All enrolled SSc patients never received nintedanib treatment. Finally, five age matched voluntary healthy subjects (HSs) were included into the study.
Isolation of monocytes, differentiation into MDMs and treatmentVenous blood sample (20mL) was collected from all enrolled HSs, SSc patients no-ILD and SSc-ILD patients, and peripheral blood mononuclear cells (PBMCs) were isolated through density gradient centrifugation using Ficoll-Paque, in accordance with the manufacturer’s protocol (Sigma-Aldrich, Milan, Italy). Monocytes were isolated from PBMCs using the EasySep human monocyte enrichment kit without CD16 depletion (Stemcell Technologies, Vancouver, Canada), and their viability and purity were assessed by Flow Cytometry.
The isolated monocytes were plated in tissue culture dishes (Eppendorf, Hamburg, Germany) at the concentration of 1.5 × 106 cells and then stimulated with phorbol myristate acetate (PMA, 5 ng/mL) in growth medium (RPMI with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% L-glutamine, Euroclone, Milan, Italy) for 24 h to induce their differentiation into MDMs.
Subsequently, only cultured MDMs obtained from SSc-ILD patients were subjected to the treatment with nintedanib as follows: a part of cultured MDMs was maintained in growth medium without any treatment (untreated cells), another part was treated with nintedanib (Boehringer Ingelheim GmbH & Co. KG, Biberach, Germany) at the concentration of 0.1µM, and another part was treated with nintedanib at the concentration of 1µM for 3, 16, and 24 h.
Cultured MDMs obtained from HSs as well as from SSc patients no-ILD were maintained in growth medium without any treatment (untreated cells) for 24 h.
At the end of the treatment, conditioned medium was collected to investigate the level of the active form of TGFβ1 (as a specific profibrotic M2 cytokine), whereas cultured MDMs were lysed to isolate both RNA and proteins for the evaluation of gene and protein expression of specific M2 cell surface phenotype (CD204, CD163, CD206) and functional markers (MerTK, IL10, TGFβ1) using quantitative real-time polymerase chain reaction (qRT-PCR) and Western blotting. The in vitro experiments were performed on cultured MDMs obtained from each enrolled SSc-ILD patient, obtaining a total number of ten independent in vitro experiments. The experimental design was represented in Fig. 1.
Fig. 1
Study design of in vitro experiments with cultured MDMs obtained from SSc-ILD patients. After collecting a venous blood sample and isolating peripheral blood mononuclear cells through density gradient centrifugation using Ficoll-Paque, human monocytes were extracted using an isolation kit and then stimulated to induce differentiation in monocyte-derived macrophages (MDMs). An aliquot of cultured MDMs isolated from each enrolled SSc-ILD patients was subsequently maintained in growth medium (untreated), another aliquot was treated with nintedanib at concentrations of 0.1µM and another aliquot was treated with nintedanib at the concentration of 1µM for 3, 16, 24 h. PBMCs: peripheral blood mononuclear cells; PMA: phorbol myristate acetate; MDMs: monocyte-derived macrophages. The picture was created with BioRender.com
Quantitative real-time polymerase chain reaction (qRT-PCR)RNA was isolated using the RNA/Protein Purification Plus kit (Norgen Biotech, Thorold, Canada) and quantified by nanodrop to assess its integrity. For each experimental condition, first-strand complementary DNA (cDNA) was synthesized from 1 µg of total RNA using QuantiTect Reverse Transcription Kit (Qiagen, Milan, Italy). The qRT-PCR was performed on an Eppendorf Realplex 4 mastercycler (Eppendorf) using a SYBR green mastermix detection system in a total volume of 10 µl, loaded in triplicate.
M2 phenotype markers were investigated using the specific primers for human CD204 (NM_002445), CD206 (NM_002438), CD163 (NM_004244), as cell membrane markers, and primers for human MerTK (NM_006343), TGFβ1 (NM_000660) and IL10 (NM_000572), as functional markers. Primers for human β-actin (NM_001101) was used as housekeeping gene.
Gene expression values have been calculated using the comparative ΔΔ cycle threshold (ΔΔCT) method and corresponded to the expression level (fold-increase) of the target gene compared to untreated cells [28]. The melting curve was included in all qRT-PCR assays to confirm the specificity of the SYBR green assay.
Western blotting and related densitometric analysisProteins were isolated using the RNA/Protein Purification Plus kit (Norgen Biotech) and quantified by Bradford method. Subsequently, 20 µg of protein was separated via electrophoresis on pre-cast 4–20% gradient tris-glycine gels (GenScript, Piscataway, USA) and then transferred onto a nitrocellulose membrane (Bio-Rad, Milan, Italy). After 1 h in blocking solution, membranes were incubated overnight at 4 °C with primary antibodies against human CD204 (dilution 1:400, Santa Cruz Biotechnologies, Dallas, Texas, USA) CD206, CD163, MerTK (dilution 1:500, Cell Signaling Technology, Danvers, Massachusetts, USA) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, dilution 1:1,000; Santa Cruz Biotechnologies). Membranes were subsequently incubated with specific horseradish peroxidase (HRP)-conjugated secondary antibodies (dilution 1:2,000; Cell Signaling Technology) for 1 h at room temperature.
Protein synthesis was detected using an enhanced chemiluminescence system (SuperSignal West Pico PLUS Chemiluminescent Substrate, Thermo Scientific, Rockford, USA) and the densitometric analysis was performed by the UVITEC Image Analysis System (UVITEC, Cambridge, UK).
For each experimental condition, the value of the protein synthesis of the investigated molecules was normalized to that of the GAPDH as housekeeping protein. The resulting value of each treatment was then normalized to that of the corresponding untreated cells (considered as the unit value).
Enzyme-linked immunosorbent assay (ELISA)Cell supernatants were collected and stored at -80 °C. The endogenous level of the active form of TGFβ1 was determined by ELISA assay using Ella automated immunoassay system, in accordance with the manufacturer’s protocol (Bio-techne, Minneapolis, Minnesota, USA).
In the first phase of the procedure (sample activation step), 80µL of sample was mixed with 20µL of 1 N HCl and incubated for 10 min at room temperature to acidify the sample. At the end of incubation, the acidification of samples was neutralized by adding 20µL of 1.2 N NaOH/0.5 M HEPES solution to obtain an activated cell culture supernatant, which was subsequently diluted 5 folds with a specific sample diluent (20µL of activated sample was added to 80µL of sample diluent). At the end of this phase, 25µL of each diluted sample was transferred into the TGFβ1 Ella cartridge.
The levels of TGFβ1 in the conditioned medium were expressed as pg/mL.
Statistical analysisStatistical analysis was carried out by non-parametric Mann–Whitney U test using GraphPad Prism (version 8.4.0, GraphPad Software, San Diego, CA, USA) to compare gene and protein expression of investigated M2 markers between cultured MDMs obtained from HSs, SSc patients no-ILD, and SSc-ILD patients. Moreover, non-parametric Wilcoxon test was used to compare paired treatments of each in vitro experiment with cultured MDMs obtained from SSc-ILD patients and treated with nintedanib. Any p value lower than 0.05 has been considered as statistically significant. Results of qRT-PCR and Western blotting were analyzed and graphically reported as median with range.
留言 (0)