Cell lines and reagents
The prostate cancer adenocarcinoma cell line PC-3 (catalog no. CRL-1435; ATCC) and prostate carcinoma cell line LNCaP (catalog no. CRL-1740; ATCC) were obtained from ATCC, Egypt. The cells were cultured in RPMI-1640 complete growth medium (catalog no. 12633-012; Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (catalog no. 098105; Multicell). The seeded cells were incubated at 37 °C and 5% CO2 in a humidified incubator, and the medium was changed every 48 h. Once the cells reached 80% confluency, they were dissociated using 0.25% trypsin (catalog no. 15400-054; Gibco) and plated in 24-well cell culture plates at a density of 70,000 cells per well for 24 h before treatment.
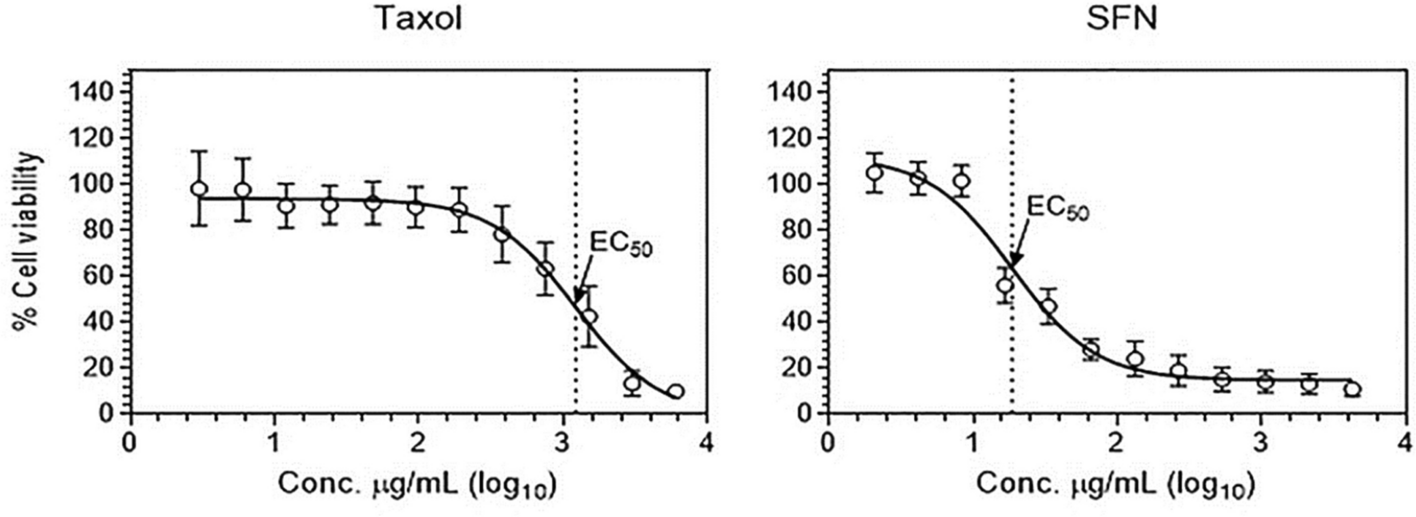
Paclitaxel (PTX) (catalog no. T7402; Millipore Sigma) was dissolved in dimethyl sulfoxide (DMSO) (catalog no. d5879; Sigma-Aldrich) at a concentration of 50 mg/mL. Sulforaphane (SFN) (catalog no. s4441; Millipore-Sigma) was diluted in DMSO to a concentration of 5 mg/mL. Different concentrations of PTX or SFN were freshly prepared in a complete culture medium before treatment.
Cell viability assay
To examine the effect of PTX and SFN on PC-3 cell viability, cells were grown in complete medium in 48-well plates at a density of 35,000 cells per well for 24 h before treatment. To investigate the effect of PTX in combination with SFN on PC-3 cell viability, we dissolved both drugs at equal concentrations starting at 100 ng/ml to 2500 µg/ml. Thiazolyl blue tetrazolium bromide (MTT) (catalog no. m-5655; Sigma) was used for the cell viability assay. MTT was dissolved in phosphate-buffered saline at a concentration of 5 mg/mL. After treating the cells with PTX, SFN, or PTX + SFN, MTT was added to each well of 24-well plates at a final concentration of 1 mg/mL directly to the culture medium. The plates were then incubated at 37 °C for three hours.
The culture medium was then removed, and the MTT formazan crystals were dissolved in 500 µL of MTT solvent (4 mM HCL, catalog no. acs393; BDH, 0.1% Nonidet P40, catalog no. 74,385; Fluka, in isopropyl alcohol, catalog no. un1219; Omnisolv) on a rocker in the dark for 15 min.
Then, 100 µL of the dissolved MTT crystals were transferred to each well in a 96-well plate and were read on a SPECTRAmax PLUS384 Microplate spectrophotometer set to a 590 nm wavelength. The absorbance values were used to calculate the percentage of viable cells relative to the untreated control cells.
Cell lysate preparation, total protein quantification, and Western blot analysis
After the treatments, the culture medium was removed, and cells were harvested in Radioimmunoprecipitation assay buffer (RIPA) containing a 1x protease inhibitor cocktail (catalog no. PI-78439c; Thermo Scientific). Total protein quantification was conducted using the BioRad protein assay (catalog no. 500-0006; BioRad) following the manufacturer’s protocol.
Total protein was denatured by adding 2x Laemmli buffer (SDS, 4%; β-mercaptoethanol, 10%; glycerol, 20%; bromophenol blue, 0.004%; Tris-HCl, 0.125 M) in a 1:1 (v/v) ratio and boiled at 95 °C for 5 min. Then, 50 µg of protein per sample was loaded into 10% SDS-PAGE. BLUelf pre-stained protein ladder (catalog no. PM008-0500; Frogga Bio) was used as a molecular weight marker (5-245 kDa).
The separated protein bands were transferred to a nitrocellulose membrane (catalog no. rpn203D; EG Healthcare). The membranes were immune-probed with rabbit polyclonal anti-caspase-3 (catalog no. AAP-113E; Stressgen), rabbit monoclonal anti-Bax (catalog no. ab32503; Abcam), rabbit monoclonal anti-Bcl2 (catalog no. ab32124; Abcam), and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (catalog no. 4699–9555; Biogenesis).
To detect the immune-probed protein bands, we used peroxidase-affiniPure goat anti-mouse IgG (catalog no. 115-035-003; Jackson ImmunoResearch) or peroxidase-conjugated goat anti-rabbit IgG (catalog no. 111-035-003) as secondary antibodies. Band visualization and densitometric analysis were carried out using Pierce ECL Western Blotting Substrate (catalog no. PI-32,106; Thermo Fisher Scientific), Chemi Doc XRS system, and Image Lab 6.0 software (BioRad).
Fluorescent microscopy and image analysis
Cells were grown on coverslips placed at the bottom of each well in 24-well plates at a density of 70,000 cells per well for 24 h before treatment.
Following treatment, the cells were fixed in ice-cold methanol (catalog no. a412; Fisher Chemicals) for 10 min at -20 °C. Methanol treatment permeabilized the cell membrane and allowed 4’,6-diamidino-2-phenylindole (DAPI, catalog no. d21490; Molecular Probes) to penetrate and stain the nuclear chromatin. DAPI was prepared in phosphate-buffered saline (PBS) at a concentration of 300 nM and added to the cell monolayers for 5 min, followed by three washes (5 min each) in PBS.
Cells were then mounted using prolonged gold anti-fade reagent (catalog no. p36930; Invitrogen) and visualized with confocal microscopy (Zeiss, Oberkochen, Germany) using ZEN 2012 software. Images were acquired using appropriate filter settings and were analyzed using ImageJ software for the quantification of nuclear DAPI staining. The number of fluorescently stained nuclei was counted per field of view, and the average number of stained nuclei was calculated.
Propidium iodide (PI) staining and cell cycle analysis
After treatment, PC-3 cells were fixed for 30 min in 70% ethanol at 4 °C. The cells were then washed twice in PBS, and 100 µg/ml of RNase A (catalog no. 1,007,885; Qiagen) was added, followed by incubation for 20 min at 37 °C. The cells were then washed twice with PBS. Next, the cells were incubated in 3 µM PI (catalog no. P4170; Sigma) in staining buffer (100 mM Tris, pH 7.4, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2, 0.1% Nonidet P40) for 15 min at room temperature. Cell cycle analysis was carried out using a flow cytometer (Guava® easyCyte; Millipore Sigma). Data acquisition and analysis were carried out using Guava cell cycle data acquisition and analysis software (Guava Technologies).
Annexin V and PI dual staining and flow cytometry analysis
PC-3 cells were harvested and washed twice with ice-cold phosphate-buffered saline (PBS). The cells were then resuspended in annexin V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) at a concentration of 1 × 10^6 cells/ml. Annexin V-FITC conjugated (catalog no. A13199; Thermo Fisher) was added at a dilution of 1:100, and the cells were incubated in the dark for 15 min at room temperature.
Following Annexin V staining, the cells were washed twice with the Annexin V binding buffer and resuspended in the same buffer. PI (final concentration of 3 µM) was added, and the cells were incubated in the dark for 15 min at room temperature. After PI staining, the cells were washed twice with the annexin V binding buffer and fixed in 1% formaldehyde prepared in the Annexin V binding buffer for 10 min on ice. The fixed cells were then washed twice with PBS, and RNase A was added at a final concentration of 100 µg/ml. The cells were incubated for 20 min at 37 °C to digest RNA.
Prior to flow cytometry analysis, the cells were washed twice with PBS and resuspended in 500 µL of PBS. Annexin V/PI positivity and data analysis was conducted using a flow cytometer (Guava® easyCyte; MilliporeSigma) and Guava data acquisition and analysis software (Guava Technologies).
Cell populations were classified into four categories based on the staining pattern: viable cells (Annexin V-/PI-), early apoptotic cells (Annexin V+/PI-), late apoptotic or necrotic cells (Annexin V+/PI+), and necrotic cells (Annexin V-/PI+). The percentage of cells in each population was determined by gating on the appropriate regions of the Annexin V/PI dot plot.
Statistical analysis
Data were obtained from n independent biological experiments and are presented as the mean of individual values with standard deviation (SD) error bars or as box-and-whisker plots showing the median, the 25th and 75th quartiles, as well as the minimum and maximum values.
To evaluate synergistic effects, the methodology established by Slinker et al. [24] was employed. Synergy was determined based on the criterion that the combined treatment effect (PTX + SFN) should surpass the cumulative effect of the individual drugs, as indicated by the equation:
$$\left[\mathrm\;\left(\mathrm+\mathrm\right)\;>\;\mathrm\;\left(\mathrm\right)\;+\;\mathrm\;\left(\mathrm\right)\right]$$
Western blot densitometric analysis was carried out using the ChemiDoc XRS system, Image Lab 6.0 (Bio-Rad), and TotalLab TL120. GraphPad Prism 6 and Microsoft Excel software were used for statistical analyses and graph generation.
Student’s t-test was used to compare two groups, and one-way analysis of variance (ANOVA) with Tukey’s correction was used for multiple comparisons to determine the significant difference between PTX, SFN, PTX + SFN, and the PC-3 non-stimulated cells (NS) that had not received any therapies.



留言 (0)