
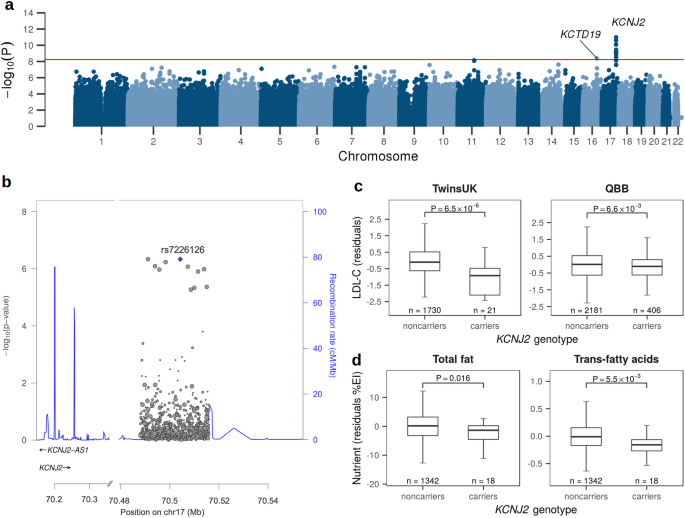
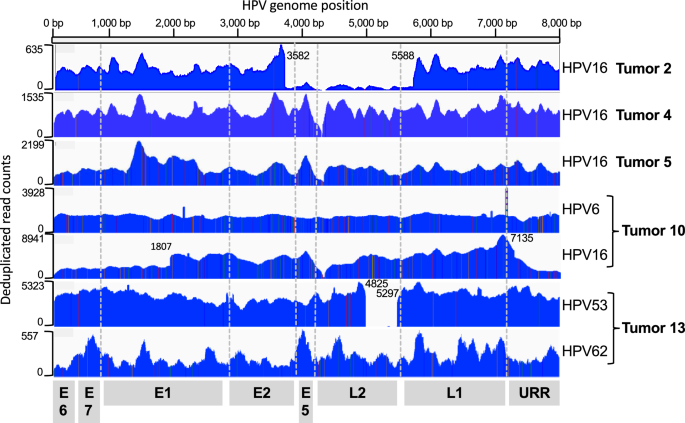
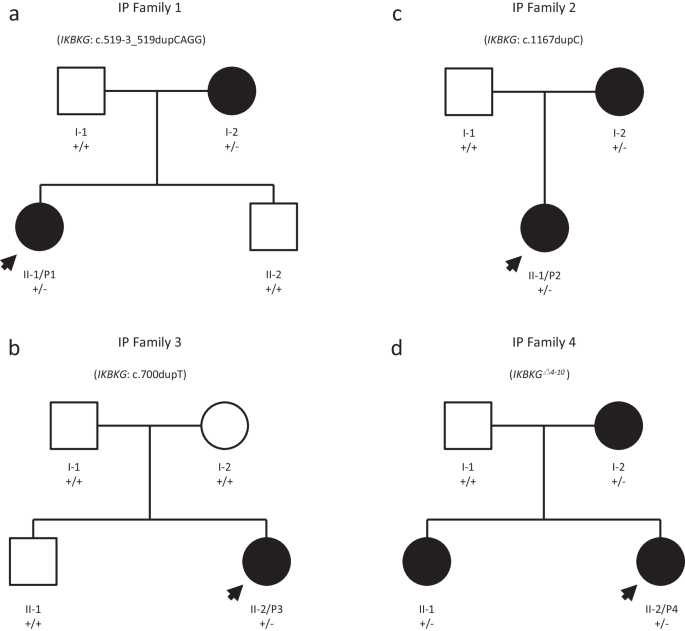
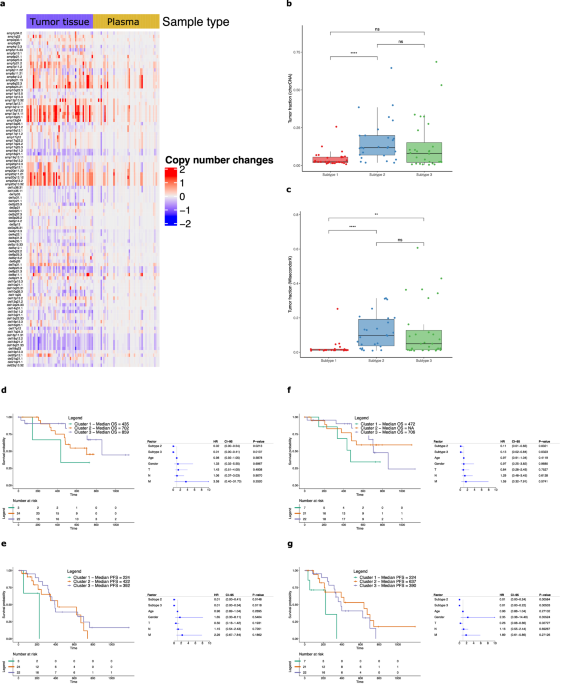
記住我
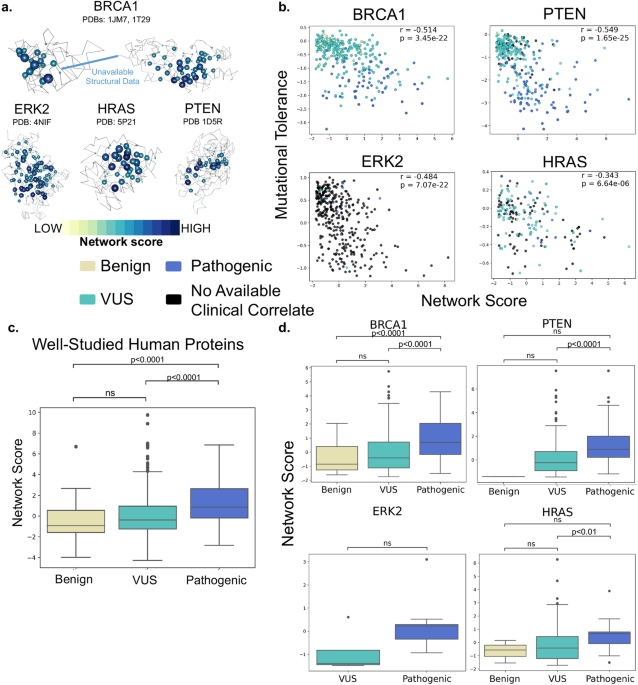



Database searches of PubMed and Dimensions yielded 4250 records plus fourteen additional references that were identified through other sources (e.g., manuscript bibliographies and hand searching of journals). A total of 269 duplicates were removed. After title and abstract screening were performed on the remaining 3995 references, 3853, records were excluded based on the inclusion and exclusion criteria (see Methods). One hundred forty-three articles underwent additional full-text screening for eligibility and 72 were excluded, including removal of case reports (n = 32). Two clinical trial cohorts (NSIGHT1 and NSIGHT2) were reported in more than one publication and were “linked” to avoid overestimating the number of unique dataset publications. Thus, a total of 71 studies were included in the evidence review (Fig. 1).
Fig. 1: PRISMA study selection flowchart.
*Reasons for exclusion included inappropriate publication type or study design (e.g., case reports), lack of primary outcome measures, and secondary publications.
Study characteristicsThe 71 studies meeting inclusion criteria were published between January 2014 to August 2022. Supplementary Tables 1–4 summarize the included studies, which are grouped into one of four categories according to healthcare setting: pediatric hospital cohorts, pediatric ambulatory cohorts, adult cohorts, and mixed cohorts.
The number of GS studies published per year gradually increased with 10 or more studies published from 2018 to 2021 (range 10–13). One or more studies were conducted in ten different countries spanning four continents (including Asia, Europe, North America, and Oceania). Forty-four percent (31/71) of studies were conducted by institutions not affiliated with the Medical Genome Initiative. Studies varied in size ranging from cohorts up to 20 patients (9/71) to studies including more than 100 patients (24/71). Combined, these studies performed GS on over 13,000 patients across diverse care settings (Fig. 2).
Fig. 2: Care setting with a breakdown of first-line GS.
Number of studies by healthcare setting, including studies where GS was the first-line genetic test (dark blue) and where GS was not the first-line genetic test (light blue).
Studies differed with respect to analysis strategy. Thirty studies (42%, 30/71) employed a panel-based initial approach based on a targeted set of established disease genes associated with the primary phenotype and variable sequential use of broader secondary gene panels or reflex to an untargeted approach. The remaining 41 studies used a phenotype-informed untargeted analysis. Phenotypic features were frequently used in the untargeted approach to provide additional prioritization of genes, but these studies did not limit the analysis to a pre-specified gene list. Additional gene-discovery analyses of candidate genes, using various methods, including investigations of rare, predicted deleterious coding variants in genes without an established human disease association, were reported in 38 studies. Analyses of non-coding variants (e.g., deep intronic, intergenic, untranslated region (UTR) variants) were conducted in 35 studies. Many non-coding analyses focused on non-coding variants present in relevant, established genes related to the phenotype or a single type of non-coding variation (most frequently deep intronic variation). A subset further limited non-coding analysis to only previously known pathogenic variants included in curated datasets (e.g., only UTR promoter or intronic variants reported in Human Gene Mutation Database (HGMD) or Clinvar)17,18,19. Other studies focused on non-coding variant analysis on specific scenarios, such as the detection of a single deleterious coding variant associated with an autosomal recessive disease gene.
Twenty-two studies used proband-only analysis, 27 studies analyzed parent-proband trios, and 22 studies included a mixture (e.g., trios with an option to include samples with only one parent available, analysis of additional affected siblings or other family members).
Fifty-nine studies applied the 2015 ACMG guidelines for variant interpretation and classification20 and the remainder of studies applied earlier guidelines21 or other systematic approaches22. Fifty-eight studies also described variants classified as variants of uncertain significance (VUS) in established disease genes. The overwhelming majority of studies reported classified or asserted pathogenic single nucleotide variants (SNVs) in known disease-causing protein-coding genes (97%, 69/71) associated with some or all of the phenotypic features. The two studies that did not report SNVs focused the analysis on copy number variants and other structural variations in a cohort of apparently balanced chromosomal anomalies23 or a cohort of undiagnosed rare disease24. Other commonly reported variants included indels (89%, 63/71) and CNVs (73%, 52/71). Less commonly reported variant types included mitochondrial DNA (mt-DNA; 20%, 14/71) variants, non-CNV structural variants (34%, 24/71), mosaic variants (11%, 8/71), and short tandem repeats including repeat expansions (14%, 10/71). Overall, the range of variant types reported increased from 2018 to 2022, with more studies reporting mt-DNA variants and non-CNV structural variants. Studies differed in the detail regarding results returned to clinicians and/or study participants which made estimates of reporting candidate genes or non-coding variants challenging, although some studies conducted such analyses. Secondary findings were reported in only 27% (19/71) of the included studies.
Seventeen studies reported turn-around time which ranged from three to 73 days. Of these 17 studies, six (35%) reported a turn-around time of 10 days or less (see Table 2).
The majority of studies were conducted in mixed patient care settings (39%, 28/71), which included a combination of ambulatory and hospital-based settings. A small number of studies reported findings in cohorts of adults (n = 7). The number of studies of pediatric hospital cohorts and pediatric ambulatory cohorts were similar (n = 16 and n = 20, respectively). Among pediatric hospital cohorts, most studies (81%, 13/16) were conducted exclusively in intensive care units and three studies included pediatric patients from intensive care units and non-intensive care unit hospital wards (Supplementary Table 1). Twelve pediatric hospital studies were restricted to infants alone.
Nearly half of the studies were performed in phenotypically heterogeneous study cohorts, where participants either had signs or symptoms suggestive of a rare genetic disorder and were not limited to a specific phenotype (e.g., epileptic encephalopathy) or single organ system (n = 34, see Fig. 3). This was followed by study cohorts composed exclusively of neurological (n = 16), cardiovascular (n = 10), ocular (n = 5), and renal and urinary tract (n = 2) disorders. Most pediatric hospital studies involved heterogeneous phenotypes (n = 14/16), with the remaining studies focused on patients with cardiovascular or neurological disorders. Of note, all seven of the studies involving adult ambulatory patients focused on organ-specific phenotypes (cardiovascular n = 4, ocular disorders n = 1, renal and genitourinary disease n = 1, neurological n = 1, immune n = 1).
Fig. 3: Phenotype categories.
Summary of indications for testing of included GS studies (total of 71 studies). Studies involving primary neurological disorders were further classified into those involving neurodevelopmental disorders (NDD) or those involving other neurological phenotypes. All cohorts in the NDD category involved individuals with developmental delay (DD) or intellectual disability (ID) and included studies where cohorts had conditions such as autism spectrum disorder or epilepsy in addition to DD/ID. Heterogeneous cohorts refer to studies where individuals had symptoms or signs concerning a genetic disorder that were not limited to a specific phenotype or single organ system.
Diagnostic yieldAcross all indications and care settings, diagnostic yield ranged from 6–86% (See Supplementary Fig. 1). The unweighted mean diagnostic yield was higher when GS was applied as a first-line genetic test (45%, range 12–73%) compared to cohorts who had previously received genetic testing (33%, range 6–86%) or in those with negative prior ES (33%, range 9–60%, see Table 3). From the random effects model of proportions, we generated a point estimate for the mean diagnostic yield across the 71 studies that accounts for cohort size and the number of diagnosed cases per cohort. This point estimate for diagnostic yield was 0.34 (95% CI 0.3012-0.3900). Diagnostic yield varied considerably across studies (tau2 0.6021, I2 93%, 95% CI 92 to 94%, see Fig. 4).
Table 3 Unweighted diagnostic yield and clinical utility based upon extent of prior genetic testingFig. 4
Comparison of diagnostic yield across studies.
Clinical utility or benefitOverall, 32% of the studies (23/71) reported on quantitative measures of clinical utility or benefit. Measures of clinical utility assessed one or more of the dimensions of utility as described by Hayeems and colleagues25: diagnostic thinking efficacy, therapeutic efficacy, patient outcome efficacy, and society efficacy. Among these studies, 70% (16/23) applied GS as a first-line test. When comparing by care setting, the clinical utility was most frequently reported in studies of hospitalized pediatric patients (14/16). Twenty-three studies reported changes in management as a result of a molecular etiology identified by GS. The proportion of patients who experienced a change in management varied among study cohorts (20–100%). Changes in management included both acute changes to treatment (e.g., medication or dietary change, other diagnostic laboratory and imaging testing) as well as long-term changes (e.g., disorder-associated surveillance). In addition to changes in management based on a new molecular diagnosis, two studies26,27 described changes in management for patients with non-diagnostic GS (7–16%). Other dimensions of clinical utility, such as patient outcome efficacy (e.g., do patients who receive GS have better outcomes than those who do not) or societal efficacy (e.g., cost-effectiveness, testing acceptable to society), were described less frequently (8/23, 9/23, respectively).
First-line GSGS was deployed as the first diagnostic genetic test in 38% (27/71) of studies. First-line GS testing was most commonly implemented in pediatric hospital-based settings (48% or 13/27 studies), whereas adult and pediatric ambulatory and mixed cohorts were more frequently reported to have undergone some genetic testing prior to GS. Most studies (56%,15/27) of first-line GS involved patients with heterogeneous phenotypes. Phenotype-specific studies of first-line GS included cardiovascular disorders (n = 3), neurological and/or neurodevelopmental disorders (n = 4), ocular disorders (n = 3), and renal and/or genitourinary tract disorders (n = 2). Clinical utility was reported in 16 studies (59%) and most of these studies (13/16) involved pediatric hospital-based settings. Changes in management associated with molecular diagnosis identified by GS were reported in all studies and varied in frequency by cohort selection and underlying molecular diagnosis (24–100%).
Study qualityAssessment of study quality by American College of Radiology (ACR) criteria28 determined that 39 studies were classified as category 1 (highest quality), 16 studies as category 2 (medium quality), and 16 studies as category 3 (modest quality). Study designs varied and included registered clinical trials, single or multi-center prospective cohort studies, and retrospective cohort studies. With the exception of the registered clinical trials, most studies provided abbreviated descriptions of inclusion and exclusion criteria for study inclusion.
Ratings roundsThe findings of this literature review were summarized and provided to the expert panel and informed the development of patient selection recommendations.
Several ratings rounds were performed with the level of consensus increasing with each round (Supplemental Results). Five core “points to consider” were developed.
Points to considerThe following recommendations were developed by a working group of the Medical Genome Initiative based on evidence from the focused literature review and expert opinion. These recommendations are intended to provide a framework for selecting patients, both pediatric and adult, with a suspected genetic disorder for first-line clinical GS testing. These recommendations are not intended to replace individual clinician judgment. As the cost of GS technology continues to decline, and additional gene-disease relationships are described, GS testing will be appropriate for a wide range of clinical indications which are not addressed here.
1.We recommend GS as the first-line genetic test for pediatric patients in intensive care units with an unexplained illness with a possible genetic etiology. Rapid GS should be considered in this setting. While there is no accepted standard definition for rapid GS, providers should consider weighing the expected time to test results with the need for time-sensitive clinical decision-making.
2.We recommend GS when sequential genetic tests are being considered because the patient’s features indicate a likely genetic cause, but do not suggest a single recognizable disorder. Examples include multiple congenital anomalies or syndromic intellectual disability when a specific disorder is not clinically identified.
3.We recommend GS when current panel testing does not encompass all the variants that are known to be causative of a disorder. Examples of variant types include coding and non-coding SNVs, small indels, CNVs, short tandem repeat expansions, copy-neutral structural variants, and mt-DNA variants. As a result, the clinician is considering sequential targeted genetic tests to comprehensively evaluate relevant variant types for the disorder in question. For example, current gene panels for intellectual disability may include coding SNVs, small indels but may not be specifically validated for the detection of certain structural variants or provide sufficient resolution to detect large CNV breakpoints.
4.We recommend GS when patients are being treated for a non-genetic condition but have a clinical course and/or response to therapy that may be better explained by a rare genetic diagnosis. Examples may include an unusually severe or prolonged clinical course, atypical response, or failure to respond to standard therapy. In addition, the clinician may wish to consider other types of genetic variation, such as pharmacogenomic alleles, that may also contribute to response to therapy and can be detected by GS.
5.We support targeted genetic testing as an alternative to first-line GS when an individual’s features strongly suggest a single recognizable genetic disorder and the clinician determines that targeted genetic testing is likely to identify the disorder. Based on the individual clinician’s judgment, the potential for phenotypically similar disorders, and the specificity of the patient’s features for the disorder in question, the clinician may still opt for a more comprehensive first-line test such as GS. Examples of targeted genetic testing include single or limited panel of genes for a specific disorder, such as FGFR3 genotyping for achondroplasia, karyotype for suspected Trisomy 21, repeat expansion testing of HTT for Huntington’s disease, panels for non-syndromic retinitis pigmentosa or hypertrophic cardiomyopathy. Current analytic capabilities of GS can detect these variant types, however, due to current resource constraints, targeted genetic testing may be a more cost-effective alternative strategy. As sequencing costs continue to decline, the relative cost-effectiveness of first-line GS for such scenarios may improve over time. Targeted testing could also be considered where features suggest a specific disorder (e.g., Lynch syndrome) and one or more of the associated genes (e.g., PMS2) occur in a region with high sequence homology that may be challenging to detect with short read GS or a greater depth of coverage is required to detect a suspected mosaic disorder. Clinicians may wish to consider the most appropriate tissue type to sequence when the specific disorder in question is known to have variable mutation load in the blood and an alternative tissue type may be preferred for detection (e.g., fibroblasts for a possible mosaic disorder or muscle for certain mitochondrial disorders associated with higher levels of heteroplasmy in muscle).
留言 (0)