
記住我



We sought to identify lncRNAs enriched within cancer cells and the TME in mCRPC based on gene expression by analyzing single-cell RNA-sequencing data from 2170 cells derived from 15 biopsies at bone, lymph node, and liver metastatic sites from 14 mCRPC patients profiled in ref. 16. We reasoned that based on the high average depth (mean depth of 1.5 million reads per cell) of sequencing coupled with the representation of various metastatic sites, treatment histories, and histologic variants, this dataset would provide substantial cell diversity and sequencing coverage for lncRNA analysis in mCRPC and the TME. Accordingly, this scRNA-seq dataset demonstrated expression of ~70% of annotated lncRNAs with lncRNAs exhibiting lower expression values when compared to protein-coding genes (Supplementary Fig. 1a, b).
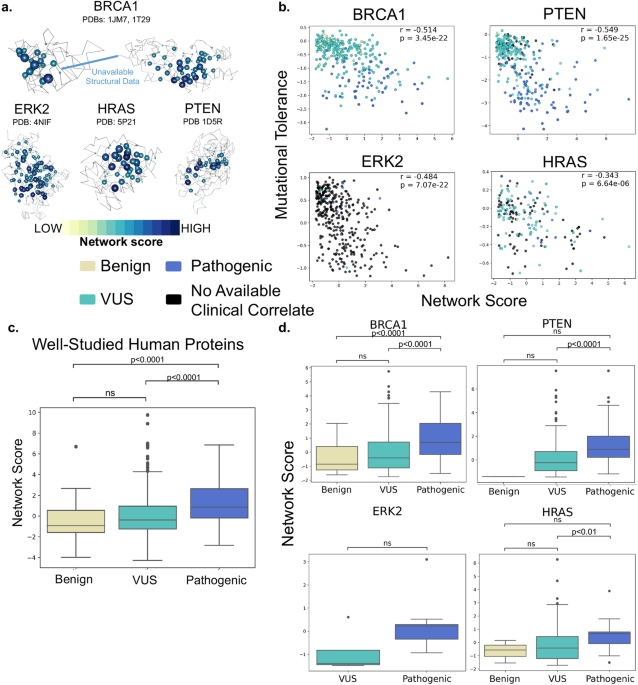
To annotate lncRNAs enriched in various cellular compartments within mCRPC, we labeled cells based on the clustering and annotation performed in the original study, which revealed clusters of prostate cancer, monocyte/macrophage, NK/T, B, neutrophil and erythroid cells. Subsequently, a computational pipeline was used to derive lncRNAs associated with each major cell type and filter lncRNAs that demonstrated low expression and poor cell specificity (“Methods”, Fig. 1a). This yielded 389 cell-enriched lncRNAs of which 91 were enriched in prostate cancer cells (pca-lncRNAs) and 298 were enriched in the TME cells (tme-lncRNAs) (Fig. 1b and Supplementary Table 1). These candidates were validated using two additional scRNA-seq datasets, one from 9 bone lesions from metastatic CRPC patients in ref. 17 and another from 11 tumors from primary prostate cancer patients in refs. 17,20. In total, 61 genes from our list of cell-enriched lncRNAs were expressed in these datasets and upon grouping of cell types, we found similar trends in cell specificity thereby validating many of these identified lncRNAs (Fig. 1c). We also compared the cell specificity of the top enriched lncRNAs with protein-coding genes within the original scRNA-seq dataset using similar criteria for cell enrichment, which demonstrated that the most enriched lncRNAs had comparable cell specificity to the most enriched protein-coding genes (Fig. 1d). We also identified lncRNAs that were known to be cell-type specific including SCHLAP1 enriched in prostate cancer cells (Fig. 1e, avg_log2FC = 4.3, FDR = 5.6 × 10−73) and SMIM25 enriched in monocyte/macrophage cells (Fig. 1e, avg_log2FC = 6.71, FDR = 1.7 × 10−137)18,21. In summary, our analysis of single-cell transcriptomic data revealed cell-enriched lncRNAs in mCRPC, validated across independent datasets, that could be used for further characterization.
Fig. 1: Discovery and validation of prostate and TME-enriched lncRNAs.
a Schematic of lncRNA discovery and validation from scRNA datasets, followed by integration with mCRPC datasets. b Heatmap of Z-scores of log2 TPM expression of prostate (91) and TME (289)-enriched lncRNAs. c Validation of common cell-enriched lncRNAs in discovery set from ref. 16 (right) with scRNA-seq datasets from ref. 17 (left) and ref. 20 (middle). lncRNAs and their corresponding clusters are listed as rows on the right side of each plot with cell types labeled as columns on the bottom and are grouped based on cell lineage. Cells in ref. 17 are grouped as such: Tumor as tumor; Mature B, mem B, Pro B, and immature B cells as B lineage; CTL1/2, NK, NKT, CD4 + /CD8+ naive, Treg Active/Inactive, and Th1/17 as NK/T cells; Mono1/2/3, TAM, TIM, mDC, Monocyte progenitor as monocyte/macrophages; and erythroid as erythroid. This dataset did not contain neutrophils. Cells in ref. 20 are grouped as such: Epithelial as tumor; B and Plasma cell as B lineage; T cell as NK/T, and Myeloid as both neutrophil and monocyte/macrophage. This dataset did not contain erythroid cells. d Top two enriched lncRNAs in each cell type vs. top two enriched protein-coding genes. e UMAP plots of SCHLAP1 enriched in prostate cancer cells and SMIM25 enriched in monocyte/macrophages.
mCRPC-associated lncRNAs have a distinct genomic and regulatory landscapeAfter identifying lncRNAs strongly associated with prostate cancer cells in mCRPC, we investigated genomic aberrations in these genes to assess the contribution of somatic mutations in lncRNA alterations. We found 13/91 (14.3%) pca-lncRNAs had at least one somatic mutation using whole-genome sequencing data from a cohort of 444 mCRPC patients in refs. 22,23,24. Most genes exhibited copy number variants (CNVs) at low frequencies (mean = 5.4%) in this cohort, with the exception of PCAT1 being amplified in 24% of patients in this study (Fig. 2a). A similar trend was noted for pca-lncRNAs in TCGA primary prostate cancer data from ref. 25 as well (Supplementary Fig. 2a). While genomic amplification could contribute to the over-expression of PCAT1 in mCRPC, studies have also attributed this to co-amplifications with the nearby oncogene MYC13. Analysis of tme-lncRNAs revealed similarly low frequencies of CNVs with the exception PVT1, which again can be attributed to its proximity with MYC amplifications (Supplementary Fig. S2b).
Fig. 2: Epigenomic features are enriched and functionally important in mCRPC-associated lncRNAs.
a Mutational landscape of pca-lncRNAs with at least one somatic mutation in mCRPC in refs. 22,23,24. b Transcription factor binding site (TFBS) motifs in pca- (n = 91) and tme- (n = 289) lncRNAs after filtering for protein-coding genes in prostate/TME cell types. c Enrichment of overlap for AR, FOXA1, and H3K27ac ChIP-seq and HMRs in prostate (n = 91), TME (n = 289), and remaining lncRNAs in hg38 (n = 16173).”*” denotes significant overlap and “ns” denotes nonsignificant overlap14,26. d Sum of methylation differences of regulatory elements in prostate, TME, and remaining lncRNAs in hg38 downsampled to 91 genes between mCRPC and benign prostate14. e Correlation of H3K27ac/HMR methylation and RNA expression in prostate (n = 28), TME (n = 27), and remaining lncRNAs in hg38 (n = 439) with statistically significant associations14.
Due to the non-focal nature of CNVs and the paucity of mutations targeting lncRNAs, we reasoned that changes in the regulatory landscape may better explain the differences in expression of cell-enriched lncRNAs. To evaluate this hypothesis, we sought to first characterize the regulatory landscape of cell-enriched lncRNAs. We first extracted promoter sequences for pca- and tme-lncRNAs and performed a differential motif search to identify putative transcription factor binding sites enriched in either prostate or TME cells, followed by filtering for transcription factor genes enriched in these respective cell types. This analysis identified several known transcription factors motifs in pca-lncRNAs and implicated in mCRPC biology, such as AR, FOXA1, NKX3.1 and the ETS family of proteins (Fig. 2b). Next, regulomic data derived from mCRPC biopsies profiled by Zhao et al.14 and Severson et al.26, such as ChIP-sequencing data for AR and FOXA1 transcription factor binding sites as well as H3K27ac peaks and hypomethylated regions (HMRs), which denote regulatory elements, was assessed for overlap with cell-enriched lncRNAs. We found a strong, significant enrichment for pca-lncRNAs (AR: 60.4%, FOXA1: 49.5%, H3K27ac: 74.7%, HMR: 75.8%,) and a weaker enrichment for tme-lncRNAs (AR: 48.3%, FOXA1: 34.2%, H3K27ac: 53.4%%, HMR: 63.8%) overlapping these regulatory features (Fig. 2c). In addition, we analyzed methylation profiles from mCRPC and benign prostate tissue for these regions near transcriptional start sites. This demonstrated a large decrease in methylation for regulatory elements in pca-lncRNAs (minimum total methylation difference = −30%) when compared to tme-lncRNAs (minimum total methylation difference = −13.3%) and a background set of lncRNAs (minimum total methylation difference = −8.65%, Fig. 2d). A similar trend was also noted for cell-specific lncRNAs between mCRPC and primary prostate cancers (Supplementary Fig. 2c). The correlation between methylation and gene expression at H3K27ac and HMR regions showed the largest anticorrelation for pca-lncRNAs (H3K27ac: mean Pearson correlation = −0.53; HMR: mean Pearson correlation = −0.55) and a weaker anticorrelation for tme-lncRNAs lncRNAs (H3K27ac: mean Pearson correlation = −0.50; HMR: mean Pearson correlation = −0.47) (Fig. 2e). Lastly, we used EpiMap data of H3K27ac profiling from various cell lines to characterize regulatory elements identified in mCRPC27. H3K27ac peaks near tme-lncRNAs showed strong enrichment in data derived from epithelial, B- and T-cell lines, which is consistent with the role for these lncRNAs in immune cells (Supplementary Fig. S2d). In contrast, peaks near pca-lncRNAs were highly enriched in epithelial, reproductive and gastrointestinal cell lines, which is a similar finding as demonstrated in prior literature (Supplementary Fig. S2d)28. Taken together, these results indicate the presence of key regulatory elements, namely transcription factor binding sites, histone acetylation peaks, and HMRs, that are enriched and functionally important for dictating gene expression primarily of pca-lncRNAs.
lncRNAs exhibit cell-specific expression changes during tumor progressionTo assess lncRNAs associated with prostate cancer progression, we performed differential expression analysis comparing bulk RNA-Seq data between primary prostate cancers in ref. 25 (TCGA, n = 333)25 and mCRPC (n = 74). Differentially expressed lncRNAs were subsequently categorized as either enriched in prostate cancer cells or TME cells based on the aforementioned single-cell analysis. This revealed several known and underappreciated lncRNAs that have significantly altered expression during tumor progression in a cell-specific fashion (Fig. 3a and Supplementary Table 2). For example, we found the pca-lncRNAs SCHLAP1 and PCAT14, both known to be enriched in prostate cancer cells, to exhibit upregulation (logFC = 1.28, FDR = 1.63 × 10−2) and downregulation (logFC = −3.77, FDR = 1.83 × 10−20), respectively, in metastases compared to primary tumors. This recapitulates similar findings for these lncRNAs in previous studies6,12. Several underrecognized associations were also detected such as an increase in NK/T-cell-enriched lncRNAs MIAT (logFC =2.1, FDR = 1.94 × 10−25) and CYTOR (logFC = 3.33, FDR = 2.86 × 10−84) in mCRPC samples. Based on further sub-setting of single cells from mCRPC biopsies, these genes were found to be enriched in CD8 + PDCD1 + T cells among NK/T-cell lncRNAs (Fig. 3b). Further supporting this, a strong positive correlation was observed in bulk sequencing data between MIAT and CYTOR expression with genes associated with a dysfunctional T-effector cell phenotype (Pearson correlation 0.66, P = 2.2 × 10−10, Fig. 3c). This result is consistent with an earlier study implicating these lncRNAs in immune infiltration within the TME29. Likewise, our study associates these genes with immune checkpoint signaling in prostate cancer due to the integration of single-cell analysis29.
Fig. 3: Single-cell analysis informs bulk expression analysis of lncRNAs associated with prostate cancer progression.
a Heatmap of differentially expressed lncRNAs in bulk RNA-seq for n = 333 primary prostate and n = 74 mCRPC samples with significantly upregulated genes in red and downregulated genes in blue25. Cell types associated with each gene are shown on the left side. Samples are ordered column-wise with normal adjacent prostate, primary prostate cancer, and mCRPC from left to right. Samples in primary prostate cancers are ordered by increasing Gleason Score and for mCRPC by the genomic status of the AR region. b Dot plot of differentially expressed NK/T-lncRNAs and T-cell subtypes in scRNA-seq data. c Correlation between expression of MIAT and CYTOR with exhausted T-cell markers from in ref. 16 in bulk mCRPC RNA-seq data16. d Methylation of differentially methylated regions (x axis) nearby differentially expressed (y axis) lncRNAs in mCRPC vs. primary prostate cancer14. e Gene plot of SCHLAP1 overlaid with HMRs (red), H3K27ac (black), AR binding sites (green), and differentially methylated regions (DMRs) (purple) between mCRPC vs. primary prostate cancer14,26.
To further explore potential mechanisms of lncRNA dysregulation during prostate cancer progression, we annotated differentially expressed lncRNAs with nearby differentially methylated regions identified in a prior report from ref. 14 between mCRPC and primary prostate cancer14. A majority of genes that were upregulated in metastases were also found to have decreased levels of CpG methylation nearby (Fig. 3d), suggesting methylation could be major mechanism of lncRNA regulation in mCRPC. Several of these included both pca-lncRNAs as well as tme-lncRNAs, suggesting that a variety of cell-enriched lncRNAs exhibit alterations in methylation as a result of tumor progression. For example, SCHLAP1 was found to contain decreased regions of methylation near regulatory elements for transcription factor binding, which may explain its upregulation in the metastatic setting (Fig. 3e). Taken together, via integration of multi-omic bulk and single-cell transcriptomic data we delineated cell-specific changes in lncRNA expression and a potential link to methylation during prostate cancer progression.
Prostate-enriched lncRNAs are associated with AR amplifications and are correlated with enzalutamide treatmentThe AR is a major driver of prostate cancer progression and treatment response and AR signaling inhibitors are commonly used for patients with mCRPC30,31. Thus, we sought to understand the role of AR signaling in lncRNA expression. We performed differential expression analysis of bulk RNA-seq data between mCRPC biopsies with AR region amplifications relative to wild-type tumors determined via whole-genome sequencing from ref. 13. This revealed several lncRNAs upregulated with AR amplifications with many of them enriched in prostate cancer cells (Fig. 4a and Supplementary Table 3). When applying this methodology to protein-coding genes in this dataset, we recovered many established AR-upregulated pca- protein-coding genes such as SLC45A3 and TMEFF2 (Supplementary Fig. 3a)14. Our results also recapitulated known AR-upregulated pca-lncRNAs such as PCAT14 (Fig. 4a)32. Moreover, this analysis yielded AR-upregulated pca-lncRNAs, such as COLCA1, which has only recently been implicated in prostate cancer33. Analysis of COLCA1 at the gene level demonstrated several AR and FOXA1 binding sites within regulatory elements near its gene body, suggesting its regulation by AR and AR co-regulators (Fig. 4b). Similarly, another example of an underappreciated lncRNAa that potentially contributes to mCRPC is TP53TG1. This pca-lncRNA was upregulated in mCRPCs with AR amplifications and was downregulated upon enzalutamide treatment. Furthermore, its promoter contains multiple regulatory elements and AR binding sites (Supplementary Fig. S3b). Prior literature has demonstrated an oncogenic role for TP53TG1 in pancreatic ductal adenocarcinoma, retinoblastoma, and nasopharyngeal carcinoma34,35,36, and future work will be needed to validate the function of this lncRNA in mCRPC.
Fig. 4: Pca-lncRNAs are associated with AR amplifications and treatment with enzalutamide.
a Volcano plot of differentially expressed lncRNAs in tumors with AR region amplifications v/s wild-type in bulk RNA-seq data from n = 64 mCRPCs16. b Gene plot of COLCA1 with HMRs (red), H3K27ac (black), AR (green) and FOXA1 (yellow) binding sites, and DMRs (purple) in mCRPC vs. benign prostate14,26. c Heatmap of AR-upregulated pca-lncRNAs in baseline and progression biopsies for matched patient samples in laser-capture microdissected (LCM) RNA-seq data13,30. d Correlation of AR-upregulated pca-lncRNAs with AR gene expression (blue) and AR activity (red) in LCM RNA-seq data30. e Expression of AR-upregulated pca-lncRNAs in LCM RNA-seq data for AR + /NE- to AR-/NE- converters (n = 3) with paired T test P value above30.
Using our list of AR-upregulated pca-lncRNAs, we next sought to validate the association of androgen signaling with pca-lncRNA expression and further explore this relationship in the context of AR directed therapies. To do this, we analyzed data from a recently published RNA-seq dataset with biopsies taken from 21 mCRPC patients at baseline and during progression with enzalutamide profiled in refs. 30,31. Although the number of lncRNAs profiled in these studies was limited, we found that expression of AR-upregulated pca-lncRNAs that were measured in this dataset, including PCAT14 and COLCA1, varied across patient biopsies with three major clusters containing either high, moderate, or low expression of these genes, respectively (Fig. 4c). These genes were subsequently found to contain positive correlations with both AR gene expression (Pearson correlation=0.4) and AR regulon activity (Pearson correlation = 0.52) as measured by the VIPER algorithm in the original studies (Fig. 4d)30. Based on this association with AR signaling, we reasoned that patients who responded to enzalutamide treatment (defined by a 50% decrease in PSA at 12 months in the original studies) should downregulate the expression of AR-upregulated pca-lncRNAs when compared to patients who failed to respond to treatment. We observed a trending decrease in pca-lncRNA expression (paired T test P value = 0.057, Supplementary Fig. 3c) and a significant decrease in AR-upregulated protein-coding gene expression (paired T test P value = 0.0017, Supplementary Fig. 3d) in patients responding to treatment.
These studies also identified three patients whose tumors developed treatment resistance due to alterations in lineage plasticity from an AR + /NE− phenotype to an AR-/NE- status, suggesting alterations in AR target gene expression. Using our methodology, expression of AR-upregulated pca-lncRNAs (paired T test P value = 0.073, Fig. 4e) showed a strong trend towards decreased expression, which was similar to the findings with AR-upregulated pca-protein-coding genes (paired T test P value = 0.22, Supplementary Fig. 3e) and the AR VIPER score (paired T test P value = 0.13, Supplementary Fig. 3f) from the original studies30, suggesting that lineage plasticity due to treatment resistance may also alter the landscape of AR-regulated lncRNAs. In summary, we found that AR amplifications are strongly associated with expression of pca-lncRNAs, which led to the identification of androgen-regulated lncRNAs. Moreover, these genes were found to be associated with response to enzalutamide and may exhibit alterations during lineage shifts as a result of treatment resistance.
TME-lncRNAs are enriched in RB1 loss tumors and are associated with poor overall survivalPrevious studies have found mutations in the tumor suppressor gene RB1 to be associated with poor overall survival across multiple tumor types and within mCRPC15,37,38,39,40. Moreover, prognostic signatures have been defined to detect genes that exhibit transcriptomic perturbations in tumors with RB1 deficiency, but these have not focused on the role of lncRNAs15. To define lncRNAs associated with RB1 loss, we performed differential expression analysis of bulk RNA-seq data between mCRPC biopsies with biallelic inactivation of RB1 relative to monoallelic and wild-type tumors determined via whole-genome sequencing from ref. 13 (Supplementary Table 4). Upon annotating lncRNAs to their respective cell types, it was found that many upregulated lncRNAs were not enriched in a particular cell type, likely reflecting non-cell-specific proliferative pathways that typically increase upon RB1 inactivation. However, a subset of upregulated lncRNAs were annotated as mostly immune cell-specific tme-lncRNAs, suggesting a link to immune infiltration in RB1 deficient tumors (Fig. 5a). In contrast, downregulated genes consisted of pca-lncRNAs associated with androgen signaling. Similar findings were found for protein-coding genes in this dataset (Supplementary Fig. 4a).
Fig. 5: TME-lncRNAs are upregulated in tumors with RB1 loss and are associated with poor outcomes.
a Differential expression analysis of lncRNAs in n = 64 bulk RNA-seq mCRPC biopsies between samples with biallelic inactivation of RB1 vs. monoallelic/wild-type RB113. Genes are highlighted based on their enrichment in cell types from single-cell analysis. b Motif enrichment for all upregulated lncRNAs (red) and tme-lncRNAs (blue) in RB1-deleted tumors. c Hallmark MSigdb pathways correlated with all upregulated lncRNAs (red) and tme-lncRNAs (blue) in RB1-deleted tumors from scRNA-seq data. d Correlation of upregulated tme-lncRNAs with the Hallmark TNFA Signaling pathway and the upregulated genes in the RBS signature from ref. 15. e Forest plots of univariate (top) and multivariate (bottom) analysis of n = 59 mCRPC samples with bulk RNA-seq for RB1 loss upregulated tme-lncRNAs and overall survival. f Forest plots of univariate (top) and multivariate (bottom) analysis of n = 59 mCRPC samples with bulk RNA-seq of RB1 loss downregulated pca-lncRNAs and overall survival.
For downstream analysis, we focused on upregulated tme-lncRNAs enriched in components of the immune system (NK/T, monocyte/macrophage, neutrophil, B lineage cells) and removed erythroid progenitor-enriched lncRNAs due to their tendency to overlap with genes associated with proliferative pathways. Differential motif analysis found that lncRNAs upregulated in RB1 loss tumors showed enrichment for E2F/SP1 transcription factors (SP1 FDR = 3.2 × 10−18, E2F4 FDR = 9.8 × 10−10), consistent with the role of RB1 in cell cycle regulation. However, immune-specific tme-lncRNAs were enriched for motifs specific to immune cells, such as the IRF family of transcription factors (IRF8 FDR = 1.6 × 10−3, Fig. 5b). Moreover, pathway analysis in scRNA-seq data revealed that this subset of genes was highly correlated with immune and inflammatory pathways (Hallmark TNFA signaling Pearson correlation=0.80) and distinct from the pathways seen with the entire gene set, which consisted of epithelial-to-m
留言 (0)