
記住我
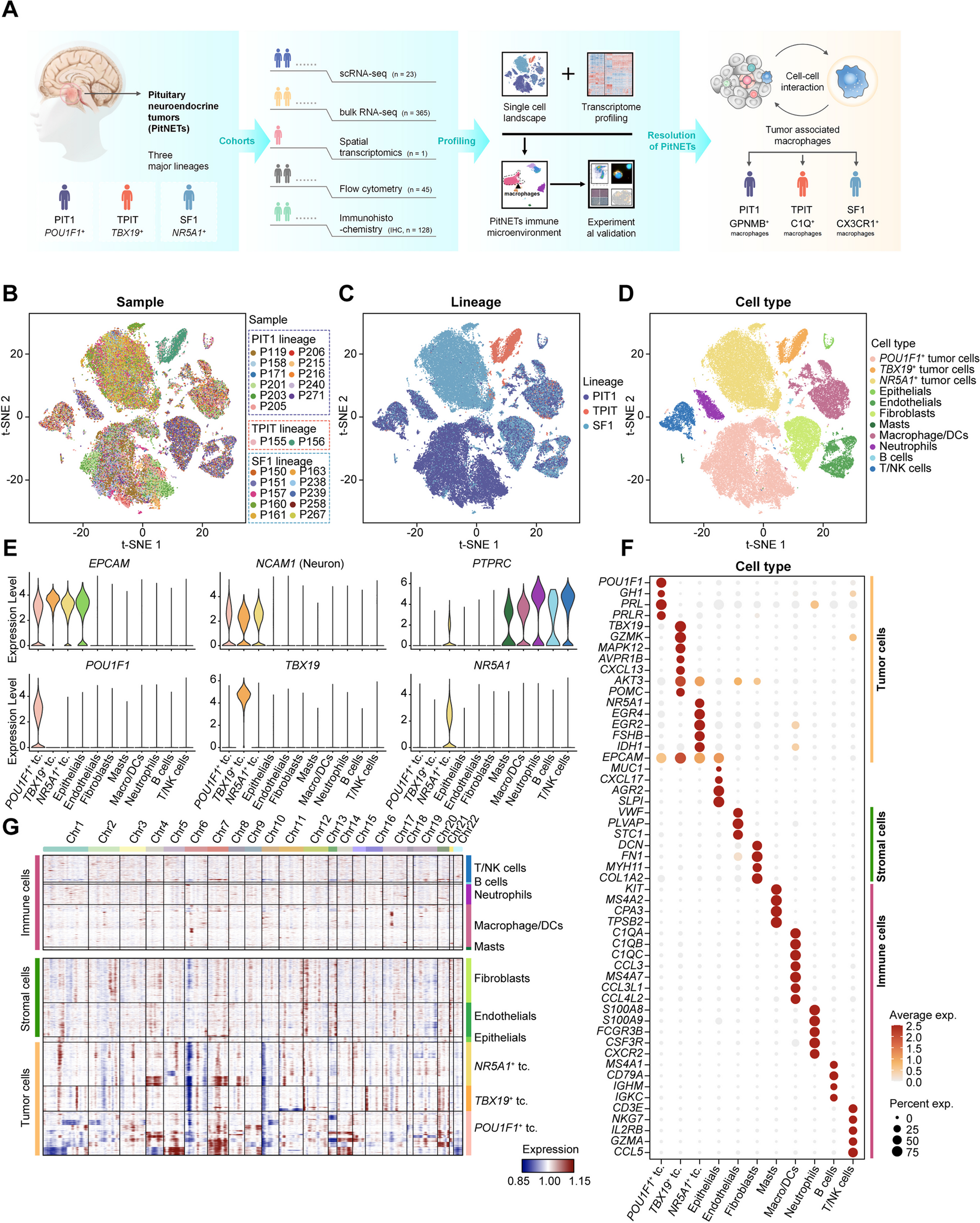
We performed whole-genome sequencing on fresh-frozen tissue samples of the prostate, seminal vesicles and lymph nodes (Table 1, Fig. 1A) to a median tumour depth of 88X using the Illumina platform (see methods for details). We systematically collected multiple samples as punches from the prostate in all cases based on histopathological evidence of tumour and from the seminal vesicles and lymph nodes where possible. We estimated cancer cell purities at 10–90% using the Battenberg algorithm (see methods). We also systematically sampled histologically normal prostate tissue punches in some patients to check for the presence of pre-malignant genomic changes. In some cases, in order to present a more complete picture of tumour spread, we augmented the data from fresh frozen samples by whole-genome sequencing of histopathologically guided sampling of formalin fixed paraffin-embedded tissue from the diagnostic archive.
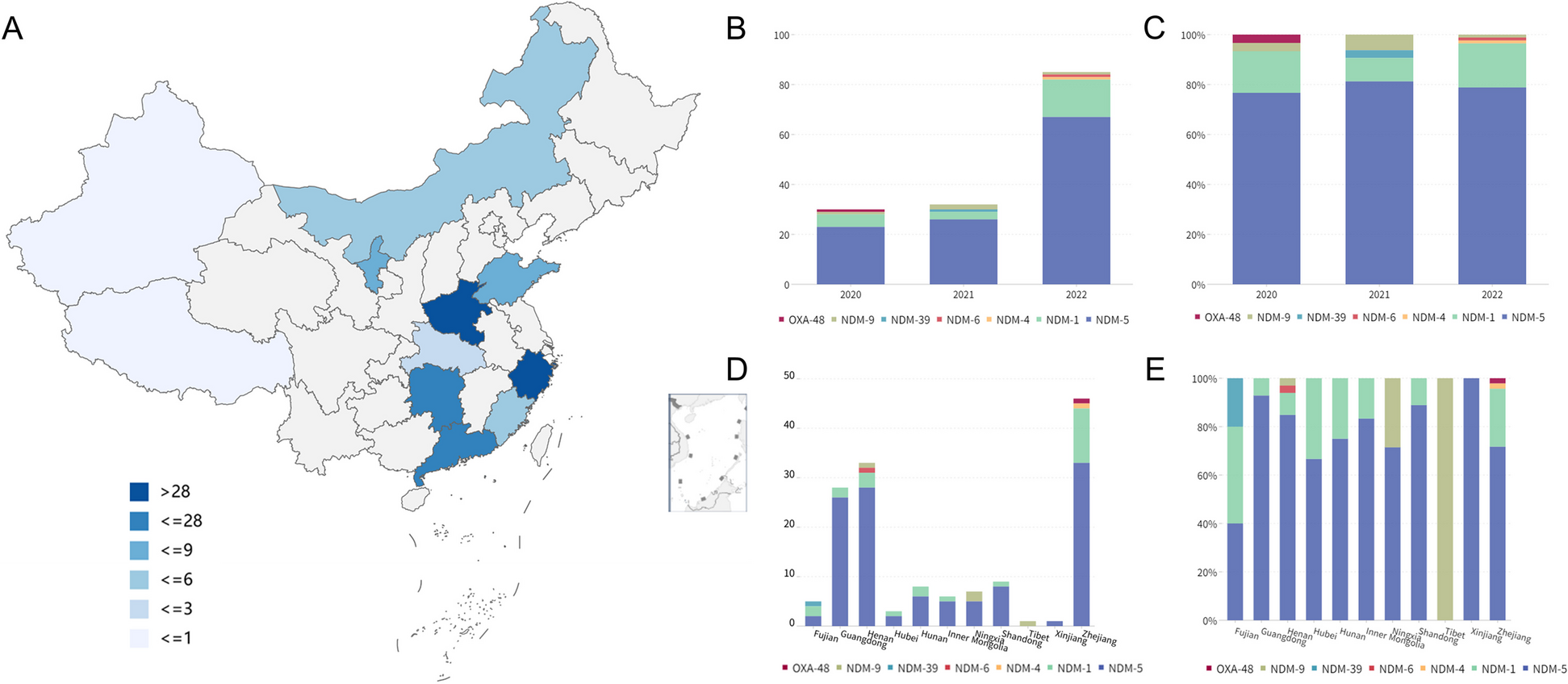
Table 1 Summary of clinical characteristics and genomic findingsFig. 1
A Schematic of sample biobanking and processing: A transverse section of the prostate was systematically sampled as 5-mm diameter × 5-mm-thick punches to obtain samples from different regions of the prostate. In addition, samples were collected from the seminal vesicles and local lymph nodes. Punches histologically confirmed to contain tumour cells by H&E staining of intermittent sections, selected histologically normal regions, seminal vesicle and lymph node samples were sequenced over the whole genome. SNVs and CNAs calculated from the WGS data were used to infer the phylogenetic structure of the tumours. B Depiction of phylogenetic trees as sunburst plots and determination of evolutionary routes: subclones identified in a sample are depicted as concentric circles/arcs, with the ancestral clone at the centre and each subsequent outer level representing a daughter clone. Multiple subclones at a given level indicate branches on the phylogenetic tree and hence that they are subclonal (cancer cell fraction < 1). Given two adjacent samples, e.g. sample 1 and sample 2, the tumour in sample 1 is inferred to give rise to sample 2 as the cancer cell fraction of clone J increased from 0.26 in the former to 1.00 in the latter
Using these spatially distinct samples, we built phylogenetic trees based on somatic small nucleotide variants (SNVs) and copy number alterations (CNAs) to assess cancer evolution in each patient. In addition to the overall phylogenetic tree for each patient, we inferred inter-sample clonal relationships based on the median cancer cell fraction of the SNVs in each clone and the spatial context of each sample. We depict the clonal composition of each sample as a sunburst plot (Fig. 1B) and the inter-sample relationships were superimposed on the histology images of the whole prostate to construct a ‘clone map’ for each patient. We present these results below.
A summary of the somatic mutations in the sequenced samples revealed that PTEN is deleted in nearly all samples across the 5 patients (Fig. 2). Further, tumour suppressor (TP53, RB1) and DNA repair (BRCA2) genes were altered in more than one individual. SNVs and CNAs were commonly present across several samples in each individual, pointing to a shared evolutionary history. Samples from #15 had the highest number of mutations affecting coding regions. We also found a higher proportion of C > T mutations in one of the four samples from #15.
Fig. 2
Oncoplot summary of SNVs and CNAs affecting known prostate cancer diver genes (Intogen) in the sequenced samples. Samples are grouped by patient ID and annotated for sample site. Integer copy numbers < 2 and > 2 were classified as deletion (Del) and amplification (Amp), respectively. Proportions of the six possible types of mutational conversions in the SNVs are shown at the bottom. Copy number alterations that were not called by Battenberg, but inferred by parsimony and supporting evidence from raw data are shown as unfilled shapes. Mutation type legend applies both to the oncoplot (middle panel) and the TMB plot (top panel) (TMB, total mutation burden; in this figure, refer to SNVs in coding regions)
Clone maps reveal evolution and spread of cancer cells within the prostateIn patient #02 (Fig. 3A, Additional file 1: Fig. S3, Additional file 1: Fig. S4, Additional file 1: Fig. S20), the LPZLat sample represents the earliest part of the malignancy, with origin of new clones as the cancer spreads along the peripheral zone to the right side. We found that the ancestral clone (clone A) comprising several copy number changes (e.g. 10q23 LOH, 16q22 LOH, 17p13 LOH) eventually gave rise to a number of subclones that were exclusively distinguished by SNVs with no further changes observed in copy number. The cancer in the seminal vesicle sample (LSV) was determined to be polyclonal (defined as a sample consisting of two or more subclones sharing a parent clone), suggesting that this is a result of either multi-clonal invasive spread or seeding from multiple metastatic events. Lymph node metastases arose from Clone A (5A_LN) and Clone J (6A_LN, 7A_LN). Clone J was also the source of the majority of the clonal heterogeneity in this patient, as it gave rise to multiple independent daughter clones (E, I, O, P). However, no copy number changes or SNVs affecting coding genes could be attributed to this clone.
Fig. 3
A Phylogenetic trees and clone maps of patient #02, B patient #08, and C patient #13, showing diverse routes of tumour evolution within the prostate, local invasion to seminal vesicles, and metastasis to lymph nodes. The phylogenetic trees are annotated with SNVs and CNAs involving known prostate cancer driver genes (Intogen). The numbers on the edges of the phylogenetic tree and the X-axis scale represent the number of SNVs assigned to the daughter clone. Metastatic events to lymph nodes are annotated on the phylogenetic tree with dashed arrows. Clone names are sorted based on cluster size, i.e. A > B > C and so on
In patient #08 (Fig. 3B, Additional file 1: Fig. S5, Additional file 1: Fig. S6), the earliest tumour location was inferred to be RPZLat and it spread along the peripheral zone to the left. The ancestral clone harboured copy number alterations involving several known driver genes (9p LOH, 10 LOH, 12p13 LOH, 13q14 LOH, 21q22 LOH). A heterozygous partial loss of the ERG gene (chr21:39868781–42856048, corresponding to exons 1–3) and the intervening region between ERG and TMPRSS2 was observed as a truncal event. TMPRSS2:ERG gene fusion was validated in all samples from this patient through analysis of the discordant and soft-clipped reads in this chromosomal region, and the breakpoints were identified as chr21:42856366 (TMPRSS2) and chr21:39867006 (ERG). Several subclonal copy number alterations were also prominent in the leaf nodes (tips) of the evolutionary tree, e.g. 12q24 LOH in clone C, and 6q LOH in clone G, affecting known prostate cancer driver genes (Intogen) such as NCOR2, FOXO3 and ARID1B. Clone D in particular harboured a loss of FOXP1 (3p13 LOH), a known tumour suppressor gene in prostate cancer [36], and notably, this clone was present in a seminal vesicle sample (LSV). Knockdown of FOXP1 in LNCaP prostate cancer cells resulted in increased migration in vitro (Additional file 1: Fig. S13). Clone H is the source of the majority of clonal heterogeneity in this patient and is the source of 6 subclones. However, no protein-coding mutations could be attributed to this clone. As in patient #02, the tumour in the seminal vesicles is polyclonal, again suggesting an invasive spread from the prostate.
In patient #13 (Fig. 3C, Additional file 1: Fig. S7, Additional file 1: Fig. S8), RLat represents the earliest region of the tumour, with the tumour spreading along the peripheral zone towards the left. Clone B is the last dominant clone (present in all intra-prostatic samples) and was the source of 3 subclones (C, D, F). Lymph node metastasis (PPF_LN) was determined to arise from clone C, although low tumour purity in the lymph node sample precluded a more detailed analysis.
Amphicrine prostate cancer arises from adenocarcinomaIn patient #10, histology of the lymph nodes revealed cancer in 12/22 nodes on the right side and 1/13 nodes on the left side. All nodal disease was amphicrine morphology (Fig. 4A). Of these, four lymph nodes were biobanked and sequenced. Amphicrine cancer is a recently described variant of prostate cancer on the spectrum of neuroendocrine differentiation, where primary cancers display features of both exocrine (classic acinar adenocarcinoma) and neuroendocrine prostate cancer [37, 38]. On standard diagnostic staining, the primary prostate cancer in this case was negative for neuroendocrine markers, but the nodal disease was positive for AR, NKX3.1 (Additional file 1: Fig. S18) and markers of NE differentiation (Synatophysin, and CD56) (Fig. 4A) with a Ki67 index of 60%. Most of the intra-prostatic malignant tumour was present towards the base of the prostate close to the seminal vesicles. Diagnostic H&E sections from this area revealed the presence of adenocarcinoma and amphicrine carcinoma in the prostate close to and in the base of the seminal vesicles with coexistence of the two histological subtypes (Fig. 4C). The nodal metastatic tumour in this case and some of the primary tumour showed extensive neuroendocrine (NE) differentiation. There was solid/nested growth with some gland formation in the primary tumour (Fig S19). Cells showed amphophilic cytoplasm, vesicular nuclei and macronucleoli, without the features of small cell or large cell NE carcinoma. The majority of the nodal metastatic disease showed solid sheets of high grade carcinoma cells with rosettes and small areas of necrosis. The morphological features were not typical of small cell NE carcinoma and were considered those of the relatively recently described entity of ‘Prostate Carcinoma with Amphicrine Features’.
Fig. 4
A Amphicrine morphology in lymph nodes, as seen by H&E staining, immunohistochemistry for Synaptophysin and CD56. B Phylogenetic tree for patient #10, with several copy number changes at the branching points. Clones that were likely sources of metastatic seeding are connected to the lymph node sample IDs by dashed arrows. C Clone map showing inferred route of cancer progression; clone J in the RLat punch expands in 17E_Ne and is associated with transformation from adenocarcinoma to amphicrine appearance
After histology-guided dissection of FFPE sections from the amphicrine and adenocarcinoma regions separately, extracted DNA was sequenced to enable a detailed reconstruction of tumour evolution in this patient. This analysis revealed that the adenocarcinoma transdifferentiated into amphicrine morphology during its spread through the prostate, with prominent genomic changes including the loss of 8p, amplification of 8q and loss of 2q22 (Additional file 1: section 5, Additional file 1: Fig. S9, Additional file 1: Fig. S10). Two seeding events were inferred, leading to metastatic growth in 614_LN/606_LN and 619_LN/854_LN lymph nodes respectively, based on SNV clustering as well as copy number analysis (Fig. 4B and C). The first seeding event to 614_LN/606_LN was inferred from the presence of a focal LOH in 9p23 (affecting the PTPRD gene); this focal LOH was not detected in the other lymph node or intra-prostatic samples. Hence, this CNA was determined to occur prior to or coincident with the emergence of clone O. Due to the features shared with 17E_Ne, and the absence of features from 17D, the seeding to 614_LN/606_LN was inferred to be between these two prostate/seminal vesicle samples. The second seeding event to 619_LN/854_LN lymph nodes was inferred by the presence of a shared CNA with 17D (12p end amplification), placing its origin chronologically after 17D. Several copy number changes (1q end LOH, 10p end LOH, 3q and 16q subclonal LOH) are also shared by 619_LN/854_LN lymph nodes, suggesting that they were coincident with the emergence of clone N. The clone map for this patient uncovered a complex 3-dimensional evolutionary history, with lymph node metastases arising from or near the seminal vesicles. Copy number analysis revealed telomeric allele imbalance in chromosomes 1q (LOH), 6p (LOH), 8p (LOH) and 12p (gain). In addition, a sample (Anterior punch) annotated as benign by histopathological assessment, but close to the main tumour was found to harbour a mutant clone with ~ 250 SNVs of a lineage distinct from the main tumour. No prostate cancer-specific driver mutations could be identified to explain the emergence of this clone and no copy number changes were detected.
Distinct histopathological features and multiple metastatic seeding events to lymph nodes in patient #15In patient #15, from whom two intra-prostatic and two lymph node samples were analysed, we were able to reconstruct the phylogenetic tree from a complex set of chromosomal gains and losses. PTEN deletion was observed in two samples (LPZMid, Iliac_LN; chr10:89074486–90215438), subclonal PTEN loss and deletion in one sample (LPZLat; chr10:89001549–90145211, 0 + 0 in 0.75 fraction subclone and 1 + 0 0.25 fraction subclone), and PTEN deletion was inferred in PPF_LN. TP53 frameshift insertion was observed in all samples from this patient. From the clonal composition and the phylogenetic tree (Fig. 5A, B, Additional file 1: Fig. S11, Additional file 1: Fig. S12), we inferred that clone H originated in LPZLat and expanded to become clonal in LPZMid sample. This clone eventually seeded metastasis to both the lymph nodes (Iliac_LN and PPF_LN) through its daughter clone A. In addition, two additional seeding events to the PPF_LN could be inferred, one event from LPZMid (clone I) and another from LPZLat (clone G). The relative timing of these seeding events could not be determined from the data.
Fig. 5
A Phylogenetic tree for #15 showing clones I, G and C as potential sources of metastatic seeding to lymph nodes. B Clone map of patient #15 showing routes of intra-prostatic spread and metastasis to lymph nodes. Clone H (pink) expands to become clonal in LPZMid. C A H&E section adjacent to the sequenced samples with areas corresponding to LPZMid and LPZLat on a plane close to the punches (superscripted with adj to denote this) showing different morphologies with D matching PSMA expression and E numbers of tumour-infiltrating lymphocyte counted by image analysis in corresponding regions compared to a distant non-tumour region
In histopathological analysis, the two intra-prostatic regions corresponding to the sequenced samples had a distinctly different morphology, with a clear demarcation between the two (Fig. 5C). The LPZMid sample had a higher Gleason score of 10 (5 + 5)/grade group 5, versus LPZLat which had a Gleason score of 7 (4 + 3)/grade group 3. There was also extensive solid pattern intraductal carcinoma adjacent to the LPZMid punch, but not LPZLat. This difference between these two samples was also reflected in the PSMA staining pattern, with minimal or absent staining in the Gleason score 10 areas around LPZTowardsMid (Fig. 5D) (H Score 10) but retained in the Gleason score 7 areas around LPZLat (H Score 100). The cancer metastases in the lymph nodes matched the morphology in LPZMid. There was also a high distribution of tumour infiltrating lymphocytes in the LPZLat sample, whereas TILs were notably less abundant in LPZMid (Fig. 5E).
Whole genome duplication and extensive chromoplexy distinguish the intra-prostatic samples in #15Whole genome duplication (WGD) is the doubling of one or both alleles of chromosomes in a usually diploid cell, resulting in triploidy or tetraploidy respectively. We considered the genomes of samples to be doubled (WGD-positive) if their ploidy was greater than 3. Hence, LPZMid, Iliac_LN and PPF_LN were found to be WGD-positive. The majority of the chromosomes have 3 or more copies, as seen in the copy number profiles (Fig. 6A). In the sample without whole genome doubling (LPZLat, ploidy = 2.80), 84% of the genome was subclonal (as opposed to 18% subclonality in LPZMid), suggesting that this sample is composed of a mixture of clones with differing copy number profiles, possibly due to a mix of WGD and non-WGD cancer cell populations. PTEN was subclonally hemizygous in this sample as seen by positive IHC staining (Fig. 6B) and copy number profiling (Fig. 6C), in contrast to LPZMid where PTEN was completely deleted. Analysis of structural variants revealed several inter-chromosomal translocation events (Fig. 6D) indicating chromoplexy and extensive chromosomal fragmentation suggestive of genomic instability in LPZLat. While several structural variants are shared between LPZLat and LPZMid (corroborating the evolutionary origin of the latter from the former), the number of inter-chromosomal translocations is higher in the former (60 vs 48). Translocations present in the PPF_LN and LPZLat samples (chr5-chr19, chr13-chr15, chr2-chr5) but not in the LPZMid or Iliac_LN samples corroborate the additional metastatic seeding event (clone G) from LPZLat to PPF_LN as inferred from SNV clustering (Fig. 5A).
Fig. 6
A Copy number plots for #15 generated by Battenberg showing a subclonal profile for LPZLat and whole genome duplication in LPZMid, IntIliac and PPFat samples. B PTEN immunohistochemistry staining corresponding to LPZLat (top) and LPZLatMid (bottom) samples and C their corresponding PTEN copy number profiles. D Structural variants plotted as circos plots, with different coloured links based on the chromosomes in which the variants’ start coordinates are located
Phylogenetic tree branches in #15 show differences in mutational signaturesUnlike other individuals, the truncal or ancestral clone in #15 was not the clone with the highest number of mutations (Additional file 1: section 6). Hence, we hypothesised that a new mutational process could have led to an increased mutation rate later in the phylogenetic tree. To investigate this further, we subdivided the SNVs into five groups based on the four sub-sections of the phylogenetic tree (GF, HA, BC, IE) and one truncal clone (D) and analysed their trinucleotide mutational signatures (Fig. 7A). While C > T mutations were the most abundant type in D and GF, this was not the case in HA, BC and IE. Deconvoluting these mutational signatures into known signatures from COSMIC v3.2, we discovered that SBS3 (associated with defective homology directed repair) increasingly contributed to the overall signature in HA, BC and IE (Fig. 7B). This suggests that there is a change in mutational processes starting at clone H. However, BRCA1 and BRCA2 genes were intact (Additional file 1: section 8: CNA profiles zoomed in on BRCA1, BRCA2, PALB2 loci, Additional file 1: Fig. S14) in LPZMid and there was no evidence of increased methylation in BRCA1 and BRCA2 promoter regions (Additional file 1: section 9: Methylation EPIC array data, Additional file 1: Fig. S15). This led us to explore alternative explanations for the increased contribution of SBS3 signature in HA, BC and IE subsections. We observed that the increased contribution of signature SBS3 coincided with whole genome duplication. We also observed increased PHH3 staining in LPZMid compared to LPZLat (Fig. 7C) indicating a higher number of cells in G2/M in the former region.
Fig. 7
A Trinucleotide mutational signatures in each subsection of the phylogenetic tree for patient #15. B Pie charts showing increasing contribution of signature 3 in tree sub-sections HA, BC and IE compared to D and GF. C Immunohistochemical staining for PHH3 (positive in G2/M phase of cell cycle), plotted as the fraction of positive cells, shows a higher proportion in the tissue region corresponding to LPZMid
Histological differences are associated with genomic changesIn order to understand the relationship between genomic changes and histology, we analysed the histological characteristics of the punches and correlated these with their corresponding clonal composition. We observed histological differences in adjacent punches in four patients (Fig. 8). In #02, prominent intraductal carcinoma (IDC) was present in RPZMid in contrast to no IDC in RTZ. Similarly, in #08 predominantly cribriform intraductal carcinoma, Gleason grade 4 + 4 was present in RPZLat whereas RLat was comprised of a higher Gleason grade tumour (4 + 5) without cribriform morphology. In #10, adjacent regions within the same FFPE section displayed features of adenocarcinoma and amphicrine carcinoma. In #15, LPZLat consisted of a Gleason grade 4 + 3 tumour and the neighbouring punch LPZMid was classified as grade 5 + 5 with no discernible glands. In all these cases, each histological subtype was found to predominantly consist of clones from a separate branch of the phylogenetic tree. We quantified the total number of SNVs uniquely present in either sample in the pair as a measure of the evolutionary distance between the two morphologies—223, 666, 324 and 4582 unique SNVs were seen in patients #02, #08, #10 and #15, respectively.
Fig. 8
Pairs of H&E-stained sections (frozen: #02, #08, #15; FFPE: #10) corresponding to sequenced regions from each of four patients (4 × magnification). Histological differences within each pair are highlighted, and the corresponding branches of the phylogenetic trees are shown in the colours of the respective leaf/tip nodes. The total number of SNVs unique to either of the two histologically distinct samples are represented as ‘Unique SNVs’. GG, Gleason grade; IDC, intraductal carcinoma of the prostate
Other general findingsWe observed a high degree of branching evolution within the prostate. In patient #02, branching occurs at nodes F, I, J and L resulting in 8 daughter clones within the prostate. In patient #08, branching at nodes H and J results in 7 daughter clones within the prostate. Intra-prostatic branching evolution was also seen to a lesser extent in #10 (at node G), #13 (at nodes A and B) and #15 (at nodes D and A).
We also observed multiple seeding events to lymph node metastases (patient #02, patient #10, patient #15). This raises the possibility that nodal metastatic potential was acquired in a clone that is not most directly associated with a seeding event. For example, in patient #02, while metastatic seeding occurs from clone A (5A_LN) and clone J (6A_LN, 7A_LN), no SNVs were observed in the coding regions of known driver genes and no new copy number alterations were acquired between clone A and J.
In the samples analysed, extensive tumour evolution occurred in the prostate, whereas fewer subclonal clusters could be identified in lymph node samples. However, in patient #10, continued evolution occurred in the lymph nodes with each of the 4 lymph nodes harbouring its own unique subclone.
In all patients except patient #10, the earliest tumour location is in the peripheral zone and the malignancy spreads circumferentially. The patterns of inferred cancer spread within the prostate suggest a predilection for the cancer cells to be limited to the prostate capsule (although there is histological evidence of extraprostatic extension, which is classified as T3a, in limited areas in all the patients in this study). In patient #10, the earliest tumour location is in the apex of the prostate, with lymph node metastases arising from the seminal vesicles.
For the majority of intra-prostatic clones in patient #02, no specific driver mutations (copy number or SNV) could be attributed. Specific driver genes can be attributed to SV invasion in #08 and lymph node metastases in #10.
留言 (0)