Limitations of Current Treatments
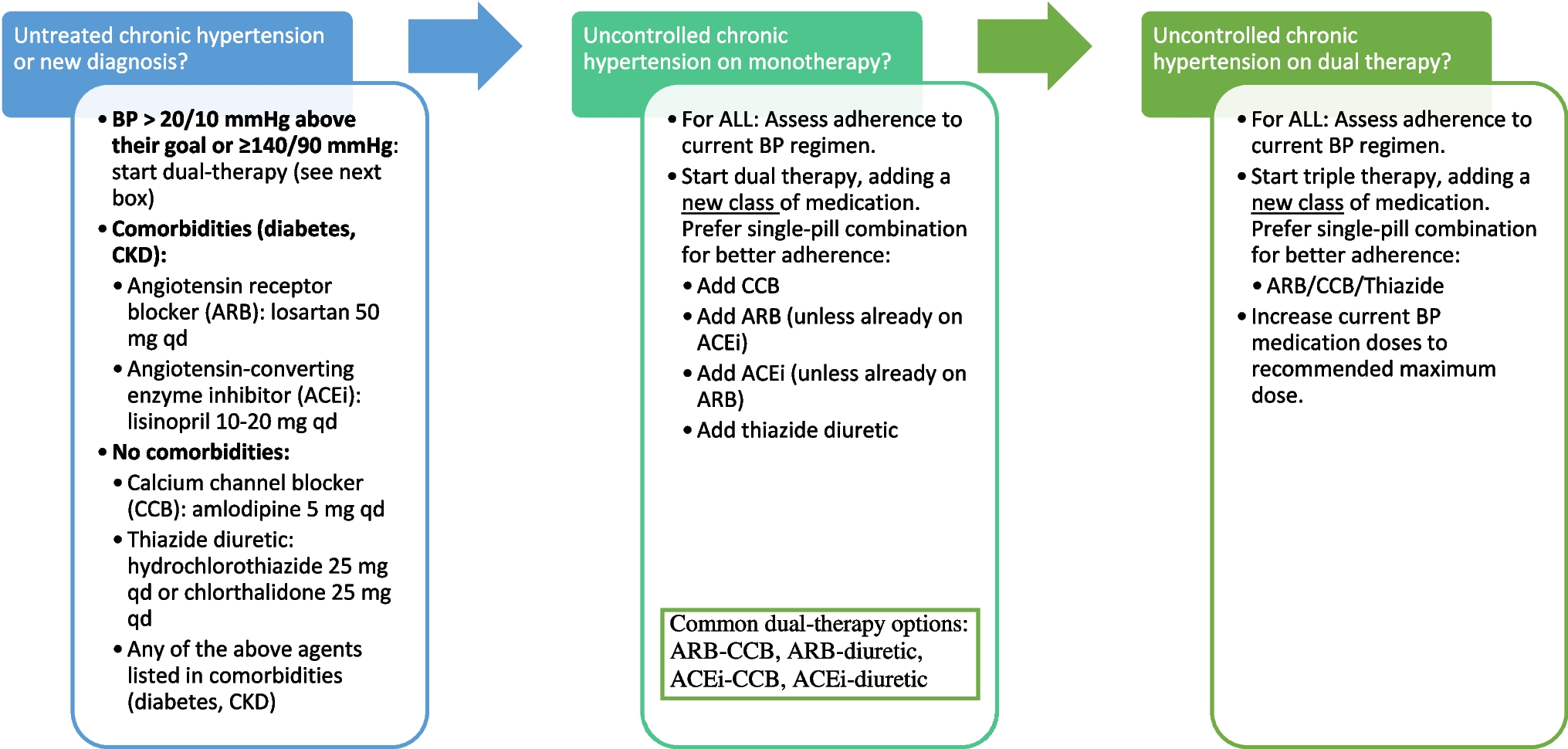
In addition to the management challenges of treatment inertia and patient nonadherence, all drug classes for hypertension have certain drawbacks. Adverse effects occur with ACE inhibitors (eg, cough, angioedema, and hyperkalemia), ARBs (eg, hyperkalemia), and CCBs (eg, peripheral edema, flushing, tachycardia, dizziness, and cardiac depression) [45]. Diuretics are also associated with adverse effects (eg, electrolyte imbalances, hyperglycemia, hyperlipidemia, and hyperuricemia); however, these are often mitigated by addition of a RAS blocker [45]. Beta-blockers can cause drowsiness, sleep disturbance, depression, visual hallucinations, erectile dysfunction, and peripheral vascular side effects [45]. Spironolactone has good efficacy and is recommended as the first choice of fourth-line agent; however, the risk of hyperkalemia precludes its use in patients with CKD and an estimated GFR < 45 mL/min/1.73 m2 or in those with serum potassium > 4.5 mEq/L [3, 46]. Treatment of OSA-associated hypertension with continuous positive airway pressure (CPAP) is shown to reduce SBP by only about 2 to 5 mm Hg, and any improvement depends on patient adherence with its use [3].
Device-based Therapies for RHT
If medical therapy is ineffective, device-based interventions may ultimately be an option for adjunctive treatment; these treatments include renal denervation (RDN) and baroreceptor stimulation [37]. The rationale for RDN is based on the role of the SNS in hypertension via renal vascular resistance, renin release, and sodium reabsorption [37]. Minimally invasive denervation can be achieved via catheter-based application of radiofrequency, ultrasound, or injection of neurotoxic agents [37]. Two RDN devices were granted breakthrough therapy designation by the US Food and Drug Administration (FDA) in December 2020 [47]. However, the magnitude of the RDN treatment effect, average BP reductions of < 10 mm Hg, suggest that this permanent approach to lowering BP will be most useful in combination with, not in lieu of, antihypertensive drugs; no devices have been approved by the FDA [3, 12, 37, 48].
Late-breaking results from a trial of radiofrequency RDN were reported at the 2022 AHA Scientific Sessions held in November 2022 [49]. The SPYRAL HTN-ON MED trial was an international, sham-controlled, randomized trial of patients with office SBP ≥ 150 to < 180 mm Hg who were stable on up to 3 antihypertensive (thiazide diuretic, CCB, ACE inhibitors, and β-blockers) medications for 6 weeks at baseline. The primary efficacy endpoint of the study evaluated changes in 24-h SBP by ambulatory BP monitoring after 6 months of study treatment, comparing RDN and sham control using a Bayesian analysis with a success threshold of 97.5%; RDN did not meet the primary efficacy endpoint (treatment difference, –0.03 mm Hg; 95% confidence interval [CI], –2.82 to 2.77). Despite the lack of significant change in 24-h ambulatory SBP and DBP, office measures of SBP and DBP were significantly decreased with RDN versus sham control (SBP, –9.9 vs –5.1 mm Hg; P = 0.001; DBP, − 5.2 vs –3.3 mm Hg; P = 0.04). The sham group also had significantly higher antihypertensive medication use during follow-up. Notably, 80% of patients had trial follow-up during the COVID-19 pandemic, and significant differences were detected when comparing 24-h ambulatory SBP, but not office SBP, in measurements collected before and during the pandemic. These differences may plausibly have affected the trial conclusions. Safety outcomes indicated a low incidence of procedure-related and clinical adverse events with RDN.
Ultrasound RDN (uRDN) has been evaluated in the multinational, randomized, single-blind, sham-controlled RADIANCE-HTN TRIO trial in patients with office measured ≥ 140 mmHg SBP and ≥ 90 mmHg DBP despite stable doses of 3 or more antihypertensives, including a diuretic [50]. At enrollment, patients were switched to fixed dose amlodipine, valsartan or olmesartan, and hydrochlorothiazide. For the primary endpoint of change from baseline to 2 months in daytime ambulatory SBP, uRDN demonstrated a significantly greater reduction than sham control (median, –8.0 mmHg [IQR, –16.4 to 0.0] vs –3.0 mmHg [IQR, –10.3 to 1.8]; median between-group difference, –4.5 mm Hg [95% CI, –8.5 to –0.3]; baseline adjusted P = 0.022). Reduction in 24-h ambulatory SBP was also significantly greater with uRDN versus sham control. Between baseline and 2 months, a similar percentage of patients in the uRDN and sham control groups had no change in their baseline antihypertensive treatment (93% vs 85%; P = 0.15). In the prespecified 6-month analysis of RADIANCE-HTN TRIO, the change from baseline in mean daytime ambulatory SBP was not significantly different between uRDN and sham control (mean difference, –2.5 mmHg [95% CI –6.7 to 1.7]; P = 0.25). The fixed-dose combination antihypertensive therapy remained unchanged at 6 months in both groups (76.9% and 82.8% in the uRDN and sham control groups, respectively); however, fewer antihypertensive medications were added in the uRDN group versus the sham control group.
Carotid baroreceptor activation therapy involves the use of an implanted pulse generator connected to leads placed next to the carotid sinus [3]. The device stimulates baroreceptors, which signal the brain to reduce sympathetic overactivity, which in turn reduces heart rate and cardiac workload, dilates the arteries, improves renal blood flow and sodium excretion, and decreases BP [3]. Baroreceptor activation therapy has been approved for the treatment of advanced heart failure [3, 51], but not as yet for use in hypertension. Nevertheless, this is a more invasive procedure than RDN that leaves the patient with internally implanted hardware.
New Pharmacologic Options
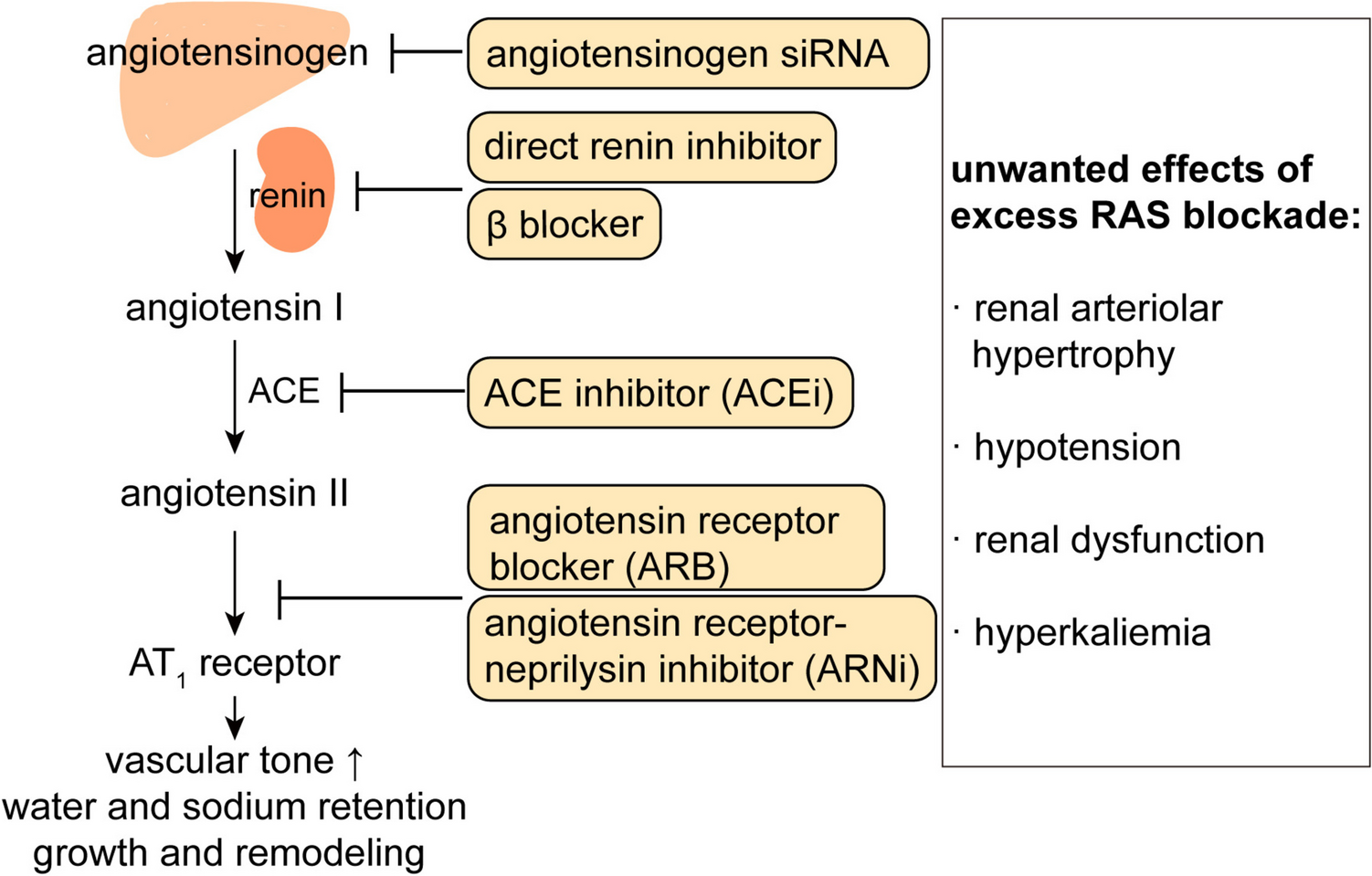
New pharmacologic options for RHT are being investigated in clinical trials, targeting both well-known disease pathways and recently elucidated pathophysiologic mechanisms (Table 1) [46]. Emerging drug classes include sodium–glucose cotransporter 2 inhibitors (eg, empagliflozin, canagliflozin, and dapagliflozin), which were approved for glycemic control in type 2 diabetes and have also been shown to reduce BP and CVD-renal events [45, 51, 52]. Neprilysin, a membrane-bound zinc endopeptidase that degrades natriuretic peptides, has been targeted in dual angiotensin receptor–neprilysin inhibitor drugs (eg, sacubitril/valsartan). This drug combination enhances natriuresis and vasodilatation and reduces BP, arterial stiffness, cardiac hypertrophy, and fibrosis [52]. Neprilysin inhibitors are also being evaluated as add-on therapy to multidrug regimens as well as in combination with other agents [46].
Table 1 Summary of recent clinical trials of investigational agents in hypertensionFiribastat is an investigational oral agent that inhibits the action of aminopeptidase A in the brain, thereby blocking conversion of angiotensin II to angiotensin III [45, 51]. Late-breaking results of the phase 3 Firibastat in Treatment-resistant Hypertension (FRESH) trial were reported at the 2022 AHA meeting [53]. Participants had uncontrolled primary hypertension despite > 80% adherence to treatment with 2 classes of drugs (for difficult-to-treat hypertension) or ≥ 3 classes including a diuretic (for RHT) and were treated with firibastat 500 mg twice daily or placebo. The primary efficacy endpoint (ie, the change from baseline in unattended automated office SBP after 12 weeks of treatment) was not met (firibastat –7.82 mm Hg vs placebo –7.85 mm Hg; P = 0.98). Secondary endpoints, including ambulatory BP monitoring, also showed no significant differences between firibastat and placebo.
Aldosterone synthase inhibitors were developed to counteract the reactive upregulation of the RAAS and SNS. The BrigHtn study, a phase 2, randomized, double-blind, placebo-controlled, multicenter dose-ranging study, evaluated BP lowering effects of the aldosterone synthase inhibitor baxdrostat [54]. Participants had a seated BP of ≥ 130/80 mm Hg and were ≥ 70% adherent to a stable regimen of ≥ 3 antihypertensive agents (including a diuretic) for ≥ 4 weeks before randomization. The primary endpoint was change from baseline in mean seated SBP at 12 weeks with baxdrostat (0.5 mg, 1 mg, or 2 mg) versus placebo. Baxdrostat showed a statistically significant reduction in placebo-corrected SBP at the 2-mg dose (–11.0 mm Hg; P < 0.001) and the 1-mg dose (–8.1 mm Hg; P = 0.003). Baxdrostat 2 mg also showed a reduction in the secondary endpoint of placebo-corrected DBP (–5.2 mm Hg). Treatment was well tolerated, with no serious adverse events considered related to the drug. Laboratory measures up to day 85 confirmed a dose-dependent reduction in 24-h urinary and serum aldosterone, an increase in plasma renin activity, and no reduction in serum cortisol, all of which support the selective mechanism of action of baxdrostat.
Other novel agents under investigation for the treatment of RHT include nonsteroidal MRA agents (eg, esaxerenone), soluble guanylate cyclase stimulators (eg, praliciguat), phosphodiesterase-5 inhibitors (eg, sildenafil), xanthine oxidase inhibitors (eg, allopurinol), dopamine beta-hydroxylase inhibitors (eg, etamicastat), recombinant B-type natriuretic peptide (nesiritide), and advanced glycation end-product breakers (eg, alagebrium) [45, 46, 52]. Vaccines targeted against angiotensin I or II may prove viable in the future as a means to decrease SBP without the need for patient adherence to complex drug regimens [46, 52]. In a phase 1 trial, the pharmacokinetic and pharmacodynamic profiles of single ascending doses of the small interfering RNA therapeutic zilebesiran, which inhibits hepatic angiotensinogen synthesis, have been evaluated in a small number of patients with hypertension [55]. In exploratory endpoint analyses, decreases from baseline in SBP and DBP were observed after 8 weeks of treatment (SBP, –22.5 [SE, 5.1] mmHg; DBP, –10.8 [SE, 2.7] mmHg). A phase 2 trial of zilebesiran in patients with hypertension is ongoing.
Endothelin Receptor Antagonists
As discussed earlier, the endothelin system of vasoconstrictor peptides plays a role in the pathophysiology of hypertension, primarily via ET-1, which was discovered to be the most potent known endogenous vasoconstrictor [23, 24, 52]. ET-1 is produced in the vascular endothelium and exerts its actions via 2 receptors, ETA and ETB [23, 24]. ETA receptors primarily mediate smooth muscle cell contraction, whereas ETB receptors have a similar function in smooth muscle but also can mediate vascular dilatation in endothelial cells through nitric oxide release [25]. Endothelin receptor antagonists (ERAs) offer either selective ETA blockade or dual blockade against both ETA and ETB receptors [24, 26]. The rationale for selective ETA blockade is to maintain the potential beneficial effects of ETB receptors [24], while the rationale for dual blockade is to suppress the function of ETA receptors plus the pathophysiologic actions of ETB receptors [24, 25]. Research has confirmed that dual receptor blockade improves vascular remodeling more effectively than selective blockade while causing fewer adverse effects [56].
Initial efforts to develop this drug class included ERA agents ambrisentan (an ETA-selective agent) and bosentan and macitentan (both dual ETA and ETB antagonists) [24]. The first ERA studied in humans was bosentan, which significantly reduced DBP versus placebo in essential hypertension, but safety concerns arose regarding hepatotoxicity [24, 26]. Ambrisentan significantly improved exercise capacity in patients with PAH compared with placebo, with adverse effects of peripheral edema, headache, and nasal congestion, but with a lower risk of hepatic injury than with bosentan [24, 57]. Macitentan was the first dual-receptor ERA to demonstrate significant decreases in morbidity and mortality among patients with PAH in a phase 3 trial, with an improved hepatic safety profile compared with bosentan and less fluid retention than with ambrisentan [24, 25]. Darusentan, a selective ETA receptor antagonist, failed to meet the primary efficacy endpoint of improvement in SBP after 14 weeks compared to placebo or guanfacine in patients with RHT and had an unfavorable safety profile [58]; further studies with darusentan were not conducted [26, 45]. Research into the ERA drug class was continued because of conflicting results regarding BP lowering, adverse effects of fluid retention, and deficiencies in trial design and patient selection in previous studies [26, 59, 60•].
Aprocitentan is an oral dual ERA that acts on both the ETA and ETB receptors and is the active metabolite of macitentan [56]. Phase 2 study results showed superior BP lowering with aprocitentan versus placebo or lisinopril in essential hypertension, leading to a pivotal phase 3 trial in patients with RHT [27]. The PRECISION trial was a blinded, randomized, phase 3 study designed to evaluate the effectiveness of aprocitentan when added to standard care for reducing BP compared with placebo over 48 weeks in patients with RHT, excluding patients with apparent or pseudo-resistant hypertension [59, 60•]. RHT was verified by a history of uncontrolled BP despite compliance with ≥ 3 antihypertensive medications of different drug classes for ≥ 1 year, SBP ≥ 140 mm Hg at screening, and no known secondary causes of hypertension [59, 60•]. The study design comprised a 4-week, double-blind, randomized treatment period with aprocitentan (25 mg or 12.5 mg) or placebo, a 32-week single-blind treatment period with aprocitentan (25 mg), and another 12-week double-blind withdrawal treatment period in which patients were rerandomized to aprocitentan (25 mg) or placebo [59, 60•]. The primary efficacy endpoint was change from baseline to week 4 in SBP measured by unattended automated office BP measurement; secondary endpoints were change in SBP from week 36 to week 40 and 24-h SBP and DBP measured by ambulatory BP monitoring at weeks 4 and 40 [59, 60•].
Results showed statistically significant and clinically meaningful reductions in SBP at week 4 in both aprocitentan dose groups versus placebo, and the primary efficacy endpoint was met [60•]. Least-squares mean (standard error) changes in SBP at 4 weeks were –15.3 (0.9) mm Hg for aprocitentan 12.5 mg, –15.2 (0.9) mm Hg for aprocitentan 25 mg, and –11.5 (0.9) mm Hg for placebo, amounting to respective differences versus placebo of –3.8 (1.3) mm Hg (97.5% CI, − 6.8 to –0.8; P = 0.0042) and –3.7 (1.3) mm Hg (–6.7 to –0.8; P = 0.0046). Least-squares mean (standard error) changes in office SBP from withdrawal baseline (week 36) to week 40 were –1.5 (0.8) mm Hg for aprocitentan 25 mg and + 4.4 (0.8) mm Hg for placebo (difference –5.8 [1.1]; 95% CI, –7.9 to –3.7; P < 0.0001). Placebo-corrected measurements from 24-h ABPM confirmed the significant reductions in SBP at 4 weeks with aprocitentan 12.5 mg (–4.2 mm Hg; 95% CI, –6.2 to –2.1) and 25 mg (–5.9 mm Hg; 95% CI, –7.9 to –3.8); reductions in DBP were comparable to those for SBP. Changes in mean 24-h ABPM measurement from withdrawal baseline (week 36) to week 40 also showed increased SBP (6.5 mm Hg; 95% CI, 4.6 to 8.5) and DBP (6.8 mm Hg; 95% CI, 5.5 to 8.0) with placebo versus aprocitentan. Reductions in BP were maintained over 48 weeks. Aprocitentan was generally well tolerated, with the primary adverse effect being edema. During the double-blind period, edema was reported by 9.1% and 18.4% of patients in the aprocitentan 12.5 mg and 25 mg groups, respectively, compared with 2.9% of patients in the placebo group; during the double-blind withdrawal period, edema was reported by 2.6% of patients receiving aprocitentan 25 mg and 1.3% of patients receiving placebo [60•]. A long-term favorable safety profile is especially important for chronic antihypertensive treatment to be used in patients who often have several comorbidities and are treated with multiple pharmacologic therapies.




留言 (0)