
記住我
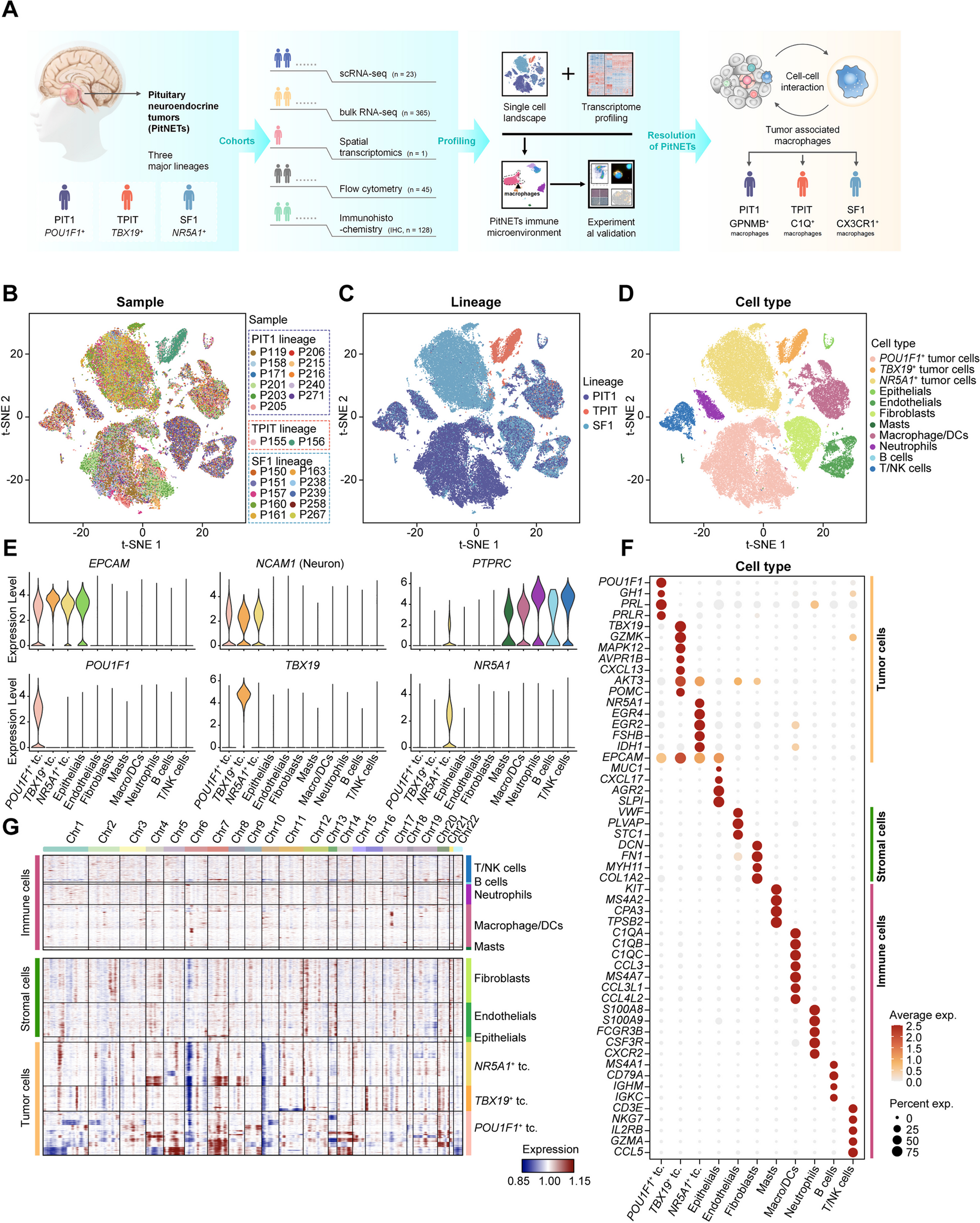
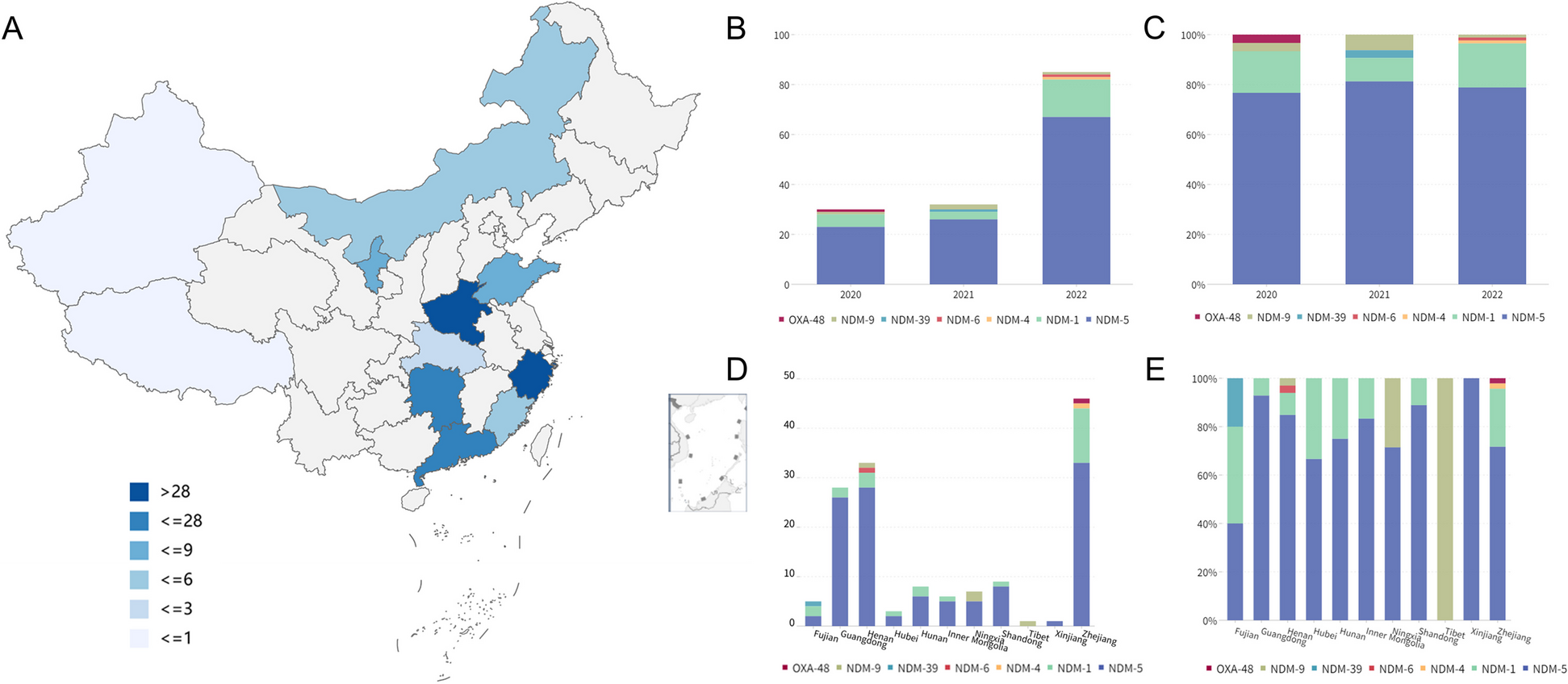
To build the atlas of cell-type-specific interactome networks, 563 patients’ scRNA-seq datasets were manually curated through literature searching (Additional file 1: Table S1). We manually filtered, collected, and uniformly processed scRNA-seq data of 44 different tumor types, including 36 solid tumor types and 8 hematological malignancies (Fig. 1, Additional file 2: Figure S3). Following strict quality control and filtration, 1,897,076 cells from solid tumors and 310,965 cells from hematological malignancies were retained. Cell counts ranged from 4590 in gastrointestinal neuroendocrine tumor to 192,889 in colorectal tumor, with a median of 26,299 per tumor type. The sample sizes ranged from 1 in cervical squamous cell carcinoma to 78 in colorectal tumor, with a median of 10 per tumor type (Additional file 2: Figure S3). For each tumor type, we integrated scRNA-seq data from different samples and performed unsupervised clustering to cluster cells into different groups. Then, we assigned cells to distinct cell types, while ensuring expression of canonical cell-type marker genes (Additional file 1: Table S4). A total of 35 different cell types were annotated in solid tumors, and 14 different cell types were annotated in hematological malignancies (Additional file 2: Figure S4).
Fig. 1
Schematic outline of the overall content is this study. The upper panel depicts the data integration and analysis workflow, including data collection, cell-type annotation, and the reconstruction of cell-type-specific interactome networks. The lower panel shows the function modules of CellNetdb, which provides versatile ways to investigate cell-type-specific interactome networks
Furthermore, the annotated CD4+ T cells, CD8+ T cells, B cells, and myeloid cells from different solid tumors were integrated with batch-effect correction to define the TIME landscape across solid tumor types. As a result, we built a large-scale pan-tumor atlas of 422,761 immune cells. Subsequently, we performed unsupervised graph-based clustering to identify various cell subsets for each immune cell type (Additional file 2: Figure S5A-D, Additional file 1: Table S5). The CD4+ T cells were clustered into seven subsets: Trm (NR4A1+, tissue-resident memory T cells), Th1 (STAT1+, T helper 1 cells), Th17 (IL23R+ and RORC+, T helper 17 cells), Treg (FOXP3+ and IL2RA+, regulatory T cells), Tn (CCR7+, naïve T cells), Tfh (PDCD1+, follicular T helper cells), and Tem (GZMK+ and CCL5+, effector memory T cells) (Additional file 2: Figure S5B). The CD8+ T cells were grouped into five subsets, including Tc17 (CCL20+, Type 17 cytotoxic T cells), Temra (KLRG1+, terminally differentiated effector memory T cells), Tn (CCR7+, naïve T cells), Tex (HAVCR2+, exhausted T cells), and Tem (GZMK+, effector memory T cells) (Additional file 2: Figure S5C). In addition, B cells and Myeloid cells were also partitioned into different subsets (Additional file 2: Figure S5A and S6D).
An atlas of cell-type-specific interactome networks across different tumor typesUtilizing the constructed single-cell atlas across 44 tumor types, we employed SCINET, a reference-guided method for inferring gene interactions from single-cell transcriptome data, to reconstruct interactome networks specific to different cell types. The reference interactome networks employed in this study include STRING, ConsensusPathDB, HumanNet, and Reactome [4,5,6, 17] (Additional file 1: Table S6). While STRING, HumanNet, and ConsensusPathDB incorporate physical protein–protein interactions as well as other sorts of interactions, such as genetic interactions, Reactome focuses primarily on reactions, pathways, and biological circuits. Both the number of nodes and number of edges in Reactome are lower than that in other networks (Additional file 2: Figure S6A-B). As a result, the scale of Reactome-guided networks is found to be lower than others when comparing cell-type-specific interactome networks estimated from different reference networks (Additional file 2: Figure S6C-D, Additional file 1: Table S8 and S9).
In addition, we applied the methodology developed by Huang et al. to compare cell-type-specific networks guided by various reference networks [30]. Two metrics, performance and performance gain, were employed to evaluate these networks. Performance was determined by the robust z-score of the true AUPRC of the gene set recovery task relative to the background of AUPRCs from the degree-matched null networks. Performance gain was computed as the difference between the AUPRC of a given network and the median AUPRC of its null networks, divided by the median AUPRC of its null networks. Specifically, we focused on the comparison of networks guided by various reference networks using the malignant cell-specific networks as an example. Tumor-associated gene sets for 16 different tumor types were sourced from the DisGeNET database (Additional file 1: Table S10). Performance and performance gain were assessed for the malignant cell-specific network of each tumor type (Additional file 2: Figure S7A-B). A strong correlation between performance and performance gain was observed (Additional file 2: Figure S7C). Our analysis revealed that cell-type-specific networks guided by STRING demonstrated the best overall performance (Additional file 2: Figure S7D).
CellNetdb accessWe have developed CellNetdb, a comprehensive data portal that facilitates the querying and visualization of cell-type-specific interactome networks (Fig. 1). To facilitate the interpretation of these networks, we have implemented versatile functional panels.
The “Taxonomy” page provides an overview of cellular taxonomy based on single-cell transcriptomes (Fig. 1). It also includes information on the cellular taxonomy of pan-tumor immune cell subsets. The use of Uniform Manifold Approximation and Projection (UMAP) allows users to visualize the 2D representation of different cell types and the expression levels of marker genes. Additionally, circle plots are utilized to visualize cell–cell communications based on aggregated signaling pathways. The number of cells for each cell type is presented in a summary table, accessible through the “Summary” panel. As an example, Fig. 2A illustrates the cellular taxonomy of lung adenocarcinoma (LUAD).
Fig. 2
The schematic features in each function module of CellNetdb. A The scatterplot represents the UMAP projection of different cell types. The hexbin scatterplot depicts the expression level of EPCAM that is a marker gene of epithelial cells. The circle plots show the cell–cell communication between different cell types. Additionally, the number of cells and marker genes for each cell type were described in a tabular form. B The graph plot depicts the local network connected to MDK gene queried from malignant epithelial cell-specific interactome networks in lung adenocarcinoma. The expression levels of all genes implicated in the local network are shown in the dot-plot heatmap where the color intensity represents the average expression level, and the size of dot represents the percentage of cells. C The somatic mutation spectra of each gene implicated in the local network of MDK is described in a table. D, E GO terms (D) and disease gene sets (E) enriched for the local network of MDK. The bar plots depict the enrichment P-values. Those genes of the corresponding GO term (D) or disease (E) implicated in the local network are labeled in purple. F The Kaplan–Meier curve depicts the difference between low- and high-expression level of SLC2A1 in TCGA lung adenocarcinoma (LUAD) cohort. The log-rank test P-value and univariate cox regression hazard ratio are labeled. SLC2A1 is implicated in the local network of MDK. G The circle plot shows the cell–cell communication between different cell types mediated by the MDK-LRP1 ligand-receptor pair in lung adenocarcinoma. The line weights represent the strengths of cell–cell communications
The “CellNet” page is designed to investigate cell-type-specific interactome networks with multifaceted function modules (Fig. 1). Users can input gene symbols after selecting the tumor type and cell type to obtain and visualize a local network containing the queried gene and its neighboring genes. Versatile function modules are employed to facilitate the understanding of biological insights. To demonstrate the resource’s usage, we focus on the MDK gene in LUAD. MDK is frequently upregulated in various human tumors and plays a crucial role in tumor development and progression [31,32,33]. By selecting lung adenocarcinoma and epithelial cells, users can enter MDK to visualize the local network of MDK (Fig. 2B). To enhance clarity, the nodes, edges, and edge weights are presented in tabular form. According to the local network, MDK exhibits strong connections to ERBB2 and ERBB3. Furthermore, the “Expression” and “Mutation” panels provide gene expression profiles and somatic mutation spectra of MDK and its neighbors, respectively (Fig. 2B,C). In terms of functional analysis, the “GO” panel indicates significant enrichment of GO terms related to cell adhesion (GO:0010811, P-value = 1.97 × 10−9), growth (GO:0040007, P-value = 3.56 × 10−8), and the ERBB2 signaling pathway (GO:0038128, P-value = 1.79 × 10−6) within the queried local network (Fig. 2D). Additionally, the local network shows significant associations with disease exacerbation (P-value = 4.72 × 10−6), malignant neoplasms (P-value = 1.12 × 10−5), and carcinoma (P-value = 4.50 × 10−5), as revealed by the “Disease” panel (Fig. 2E). By switching to the “Survival” panel, we observe that the neighboring gene SLC2A1 is associated with overall survival in LUAD patients (log-rank test P-value = 1.59 × 10−5) (Fig. 2F).
Finally, the “Communication” panel allows users to investigate how the local network influences cell–cell interactions between different cell types. We found that myeloid cells respond to malignant epithelial cells through the interaction of MDK and its receptor LRP1 (Fig. 2G). This aligns with previous study showing that MDK can interact with LRP1 to promote immunosuppressive macrophage differentiation in ERBB pathway-mutated tumors [34]. Additionally, we discover that the MDK-LRP1 pair is involved in the response of fibroblasts to malignant epithelial cells. Through interrogating the gene–gene interactions in the malignant cell-specific networks and cell–cell interactions between malignant cells and other cells, we identified several other genes may affect the MDK-LRP1-mediated intercellular crosstalk, such as ERBB2, ERBB3, HRAS, and ESR1. The ERBB pathway mutations have been shown to upregulate MDK expression in bladder cancer in previous studies. Therefore, CellNetdb can help researchers identify candidate genes that may affect intercellular crosstalk. Overall, CellNetdb aids in unraveling the cellular heterogeneity of gene–gene interactions, enhancing our understanding of the functional roles of genes within the cellular context of tumor development and progression.
Cell-type-specific network reveals topological specific genes with preferential cell-type influenceRewiring gene interactions across different cell types can lead to changes in the network topology, which may have varied functional importance depending on the cellular context. Previous studies have demonstrated that constitutive proteins can acquire context-specific effects through tissue-specific interactions [35]. To assess the functional application of cell-type-specific networks in understanding the context-specific role of genes, we utilized a metric called topological specificity (topS), introduced by SCINET [16]. This metric allows for the direct quantification of a gene’s influence in a network, beyond what is captured by connectivity and strength alone. The topS metric measures the deviation between the observed overall interaction strength of a gene within a specific cell type and the expected strength derived from a random model that preserves the network’s topology while reshuffling the cell-type-specific interaction strengths.
To illustrate the application of cell-type-specific networks in understanding the context-specific roles of genes, we focused on myeloid cells in pan-tumor TIMEs as an example. We initially investigated whether known canonical cell-type signature genes play a distinct role in the networks [16] (Additional file 1: Table S11). Our findings revealed that these signature genes exhibited significantly higher topS values in the corresponding cell subsets, indicating that the inferred local-interaction topology effectively captures context-relevant biological roles (Fig. 3A). Additionally, we identified top 500 genes for each cell subset based on their topS and tranS scores, respectively. Notably, there was a moderate concordance between the candidate genes identified by topS and tranS, even when both transcriptional and topological specificity correlate globally (Fig. 3B, Additional file 2: Figure S8), suggesting that cell-type-specific networks can complement conventional expression analysis.
Fig. 3
Topologically specific genes with cell-type-specific influence in pan-tumor myeloid cells. A The distribution of topologically specific scores (topS) is presented for each cell-type-specific network, focusing on canonical marker genes for a specific cell subset in pan-tumor myeloid cells. B Comparisons of genes prioritized by topS and tranS in various cell subsets of pan-tumor myeloid cells is depicted, with Jaccard indices calculated for the top-ranked 500 topS and tranS genes in each cell subset, and the number of shared genes displayed. C Scatterplots illustrating topologically specific genes with broad expression across different cell subsets in pan-tumor myeloid cells, with the best linear regression fit represented by the blue line and the top 5 genes with log2FC(PRtopS/PRtranS) are highlighted. The log2FC(PRtopS/PRtranS) represents log2 fold change between percentile ranks of topS and tranS
Furthermore, we identified several sets of genes that exhibited high topS scores but low tranS scores for each cell subset (Fig. 3C). These genes were found to be expressed in multiple cell subsets but had distinct topological roles across different cell-type-specific networks. Among the top 5 genes with the strongest deviation in topS relative to tranS per cell subset, we identified genes involved in generic functions such as actin binding (CAP1), protein folding (HSPA1B), and growth factor activity (MDK), as well as genes with more specific functions such as the regulation of macrophage inflammation (CD74, a cell membrane high-affinity receptor for MIF). Overall, the cell-type-specific networks enabled us to investigate genes with a preferentially influential role in a cell-type network that cannot be solely explained by their expression pattern.
Identifying prognosis-associated cell types through network connectivity analysisThe rewiring of molecular networks in diverse cell types with functional relevance can also benefit identifying disease-associated cell types. Previous studies have shown that disease genes tend to form cohesive neighborhoods in the human interactome network [36]. We proposed to deconvolve disease-associated gene sets into individual cell types through evaluating the topological properties of these genes in the cell-type-specific networks, inspired by previous studies [25]. To evaluate the statistical significance of the observed connectivity of a gene set, we employed permutation test approach by generating a null distribution by repeatedly randomly subsampling the same number of genes from the network. Therefore, by applying network connectivity analysis on cell-type-specific networks, we could potentially identify individual cell types associated with tumor patients’ prognosis. With clinical data in TCGA project, we first identified the top 500 prognostic genes based on their expression levels. We ranked these genes using the adjusted p-value of the log-rank test, which compares the overall survival of patients with high and low gene expression levels. Through connectivity analysis on different cell-type-specific networks, we found strong network connectivity for prognostic genes in non-cancerous cells, including stromal cells and immune cells (Fig. 4A), indicating that some prognostic genes may function within the non-cancerous cells of the tumor microenvironment.
Fig. 4
Cell-type deconvolution of cancer prognostic gene signatures. A Scaled within-group connectivity of top-ranked cancer prognostic gene sets in different cell-type-specific networks. Cell types presented in more than 10 cancer types were included for analysis. B Weighted degree centrality of the 128 prognostic genes in the fibroblast-specific network in gastric cancer, with emphasis on the top 10 genes. C Weighted degree centrality of genes neighboring the 128 prognostic genes in the fibroblast-specific network in gastric cancer, highlighting the top 10 genes. D Gene set enrichment analysis on the KEGG pathway for the top-ranked 30 genes neighboring the 128 prognostic genes in the fibroblast-specific network in gastric cancer. E Visualization of the network of genes neighboring ITGB1 and several focal adhesion-related genes of the fibroblast-specific network in gastric cancer, with the size of nodes representing the centrality of genes. Brown nodes denote the prognostic genes in the network. F Kaplan–Meier plot for gastric cancer patients of TCGA cohort based on the expression of ITGB1, showcasing the classification of patients into high-expression and low-expression groups by median value for analysis
Next, we focused on the strong connectivity of prognostic genes in the fibroblast-specific interactome networks in gastric cancer. Tumors can activate stromal fibroblast to become cancer-associated fibroblasts (CAFs), which then promote cancer aggressiveness [37]. We identified 128 top prognostic genes in the fibroblast-specific network, with 78 genes closely connected to each other (P-value = 0.0028). Among these prognostic genes, several were associated with the metastatic spread of tumors, such as ITGB1, CAV1, BMP4, and PDGFRB [38,39,40,41], and exhibited a high degree of centrality in the fibroblast-specific network (Fig. 4B). To gain insight into the role of these top-ranked prognostic genes in fibroblasts, we examined the top hub genes directly connected to them (Fig. 4C) and found that top 30 direct neighbor genes were enriched in estrogen signaling pathway and focal adhesion (Fig. 4D). Interestingly, several hub genes directly connected to ITGB1, which had the highest degree of centrality among the prognostic genes in the fibroblast-specific networks, included EGFR, FN1, and COL1A1 (Fig. 4E). EGFR is a member of the ErbB receptor family, known to be involved in CAFs-mediated promotion of tumor invasion and metastasis [42]. FN1, encoding fibronectin 1 derived from CAFs, can also promote invasion and metastasis [43]. COL1A1, encoding collagen type I alpha 1, has been shown to play a critical role in tumor progression [44]. Besides, the association of ITGB1 with poor survival has been corroborated by the median expression observed in TCGA-STAD samples (Fig. 4F). Thus, we propose that the prognostic effects of ITGB1 are linked to CAF-mediated tumor progression. Overall, our findings demonstrate the potential of deconvolving tumor prognostic genes into specific cell types and identifying key target genes within these cell types.
Additionally, tumors which respond well to immune checkpoint blockage (ICB) therapy often have high levels of tumor-infiltrating lymphocytes in their tumor lesions, indicating hot tumors [45]. On the other hand, tumors which do not respond well typically with low T cell infiltration, known as cold tumors [46]. Therefore, we tried to see if the connectivity of cytotoxic gene sets in the CD8+ T cells can differentiate between hot and cold tumors. We curated 159 cytotoxic genes from the Gene Ontology database and calculated their connectivity in each CD8+ T cell-specific network for different cancer types. We found that the connectivity of these cytotoxic genes is positively correlated with the proportion of CD8+ T cells in each tumor sample, serving as a potential indicator of tumor immune profile categorization into hot and cold tumors (Additional file 2: Figure S9). For example, tumor types characterized as hot, such as lung adenocarcinoma, lung squamous cell carcinoma, and head and neck squamous cell carcinoma had high levels of CD8+ T cells and connectivity of cytotoxic genes. In contrast, cold tumors like glioblastoma and pancreatic ductal adenocarcinoma had low CD8+ T cell infiltration and connectivity of cytotoxic genes (Additional file 2: Figure S9). Collectively, our findings suggest that the connectivity of cytotoxic genes may serve as a discriminatory factor in distinguishing hot and cold tumors.
Cell-type-specific interactome network commonality and difference across different tumor typesWe conducted a comparative analysis of networks specific to various cell types derived from different solid tumor types. To compare the networks, we employed two metrics based on node topological specificity and interaction strengths, respectively (see “ Methods”) [30]. Our findings suggest that networks specific to the same cell type consistently clustered together, even across different tumor types, indicating that the cellular context primarily shapes the cell-type-specific interactome networks rather than the tumor or tissue types (Additional file 2: Figure S10). Moreover, we observed that the ratio of unique edges to shared edges across tumor types was higher than that of unique nodes to shared nodes, suggesting that networks for the same cell type undergo rewiring in different tissue contexts (Additional file 2: Figure S11). Additionally, we expanded our analysis to include networks specific to various cell types derived from five hematologic malignancies, encompassing all six cell types. Like the analysis of solid tumor types, network similarity analysis revealed a similar clustering pattern (Additional file 2: Figure S12).
We retrieved and compared malignant cell-specific networks from various solid tumor types, revealing correlation blocks aligned with known physiological conditions of malignancies (Fig. 5A). Notably, malignant epithelial tumors were found to group together, and prominent correlation blocks emerged among different tumor types, such as gastric cancer, colorectal cancer, and pancreatic ductal adenocarcinoma (Fig. 5A). Application of the network community detection method Infomap identified core subnetworks enriched for functional terms related to glycolysis, metabolic processes, protein phosphorylation, MAPK signaling pathway, cell adhesion, and inflammatory response (Fig. 5B–E). The functional interpretation of these core subnetworks aligned with the properties of these tumors, such as hypoxia/Warburg effect, epithelial-to-mesenchymal transition, and tumor-associated inflammation. Hub genes with high centrality, including PTK6, FGFR3, and FGFR4, which have been recognized as therapeutic targets in tumor treatment. Additionally, MAGI3, a hub gene connected to them, has been identified as a novel substrate-binding subunit of E3 ligase, suggesting potential regulatory roles of MAGI3 associated with protein tyrosine kinases that require further verification. Overall, the core subnetworks identified from the shared interactome specific to malignant cells can provide insights into candidate tumor genes and generate testable hypotheses.
Fig. 5
Comparative analysis of interactome networks specific to malignant cells across different tumors. A The heatmap depicts the comparison of interactome networks specific to malignant cells identified from different tumor types. The red gradient panel represents the topology similarity estimated from shared nodes’ topological specificity. The green gradient panel represents the edge similarity estimated from shared edges’ interaction strengths. Network sizes are shown by number of nodes (red bars) and number of edges (blue bars). The corresponding tumor types and physiological cell types of different malignant cells are also labeled. B–E The graph plots depict four representative core subnetworks identified from the shared network of gastric cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. The centrality of each gene implicated in the subnetwork is labeled in color. The bar plots under each graph plot shows the GO (yellow bars) and KEGG (blue bars) pathway enriched for each core subnetwork
Moreover, similar patterns were observed in the comparisons of CD8+ T cell and CD4+ T cell-specific networks (Additional file 2: Figure S13 and S14). Several core subnetworks were identified for CD8+ T cell and CD4+ T cell-specific networks, respectively. Notably, a core subnetwork enriched for stress response was identified in the shared CD8+ T cell-specific network (Additional file 2: Figure S13E). These findings align with a recent study indicating that a cellular stress response state of T cells [47]. Collectively, our findings underscore the significance of considering the cellular context.
Application to prioritizing risk genesThe identification of genes involved in cancer progression presents a significant challenge with critical implications for understanding biological processes. The use of cell-type-specific networks provides a promising approach for extending gene prioritization within the context of specific cell types. Several network-based prioritization methods have been developed to rank disease-associated genes, prompting the need to determine the most suitable method. To address this, a benchmarking analysis was conducted to evaluate the performance of four representative methods—RWR [48], GenePanda [49], Node2Vec [50], and DIAMOnD [51]. Utilizing genes annotated in the Cancer Gene Census (CGC) as seed genes [52], the prioritization of risk genes was performed using malignant cell-specific networks from six different tumor types. Subsequently, the NCG 6.0 database was used as a benchmark to assess the performance of each method [53]. The results indicated that the RWR outperformed the others in prioritizing risk genes by leveraging biological networks (Additional file 2: Figure S15). As a result, the RWR method has been implemented into CellNetdb for prioritizing risk genes within the context of cell-type-specific interactome networks. The platform allows users to upload a gene list as seed genes for prioritizing risk genes within cell-type-specific networks. Case studies in uveal melanoma and acute myeloid leukemia were provided to demonstrate the utility of this platform, revealing a high proportion of candidate genes annotated as cancer drivers (Fig. 6A,B and Additional file 2: Figure S16 and S17). Additionally, genes not reported by NCG may also have roles in tumors, as demonstrated by the examples of BCL2L1, PLEKHA4, and RUNX2 [54,55,56].
Fig. 6
Prioritize risk genes in CellNetdb. A Prioritize risk genes using the interactome network specific to malignant melanocytes in uveal melanoma. The bar plot depicts the stationary probability for each top-ranked gene from random walk with restart. The reported cancer driver genes in the NCG 6.0 database are labeled in red. B Prioritize risk genes using the interactome network specific to malignant myeloid cells in acute myeloid leukemia. The reported cancer driver genes in the NCG 6.0 database are labeled in red. C Prioritize risk genes using the interactome networks specific to exhausted T cells in the pan-tumor TIMEs. D–F The expression levels of KLRD1 (D), CD27 (E), and LGALS9 (F) in six ICB therapy cohorts. The boxplots show the difference of their expression levels between ICB therapy response and non-response groups. The cohorts of different tumor types are labeled, including skin cutaneous melanoma (SKCM), clear cell renal cell carcinoma (ccRCC), and non-small cell lung carcinoma (NSCLC). The asterisks represent the degree of significance calculated using Limma (* P-value < 0.05, ** P-value < 0.01, *** P-value < 0.001). G–I The Kaplan–Meier curves of overall survival (OS) and progression-free survival (PFS) depict the differences between patients’ groups stratified by the expression level of KLRD1 (G), CD27 (H), and LGALS9 (I), respectively
Additionally, users can use interactome networks specific to different immune cell subsets to prioritize risk genes. For example, inputting a list of genes associated with T cell exhaustion as seed genes will return a list of candidate genes prioritized using the networks specific to CD8+ exhausted T cells (Fig. 6C and Additional file 2: Figure S18). Some of the top-ranked genes, such as PTPN11 (SHP2) and PTPN6 (SHP1), have been linked to T cell exhaustion [57,
留言 (0)