
記住我


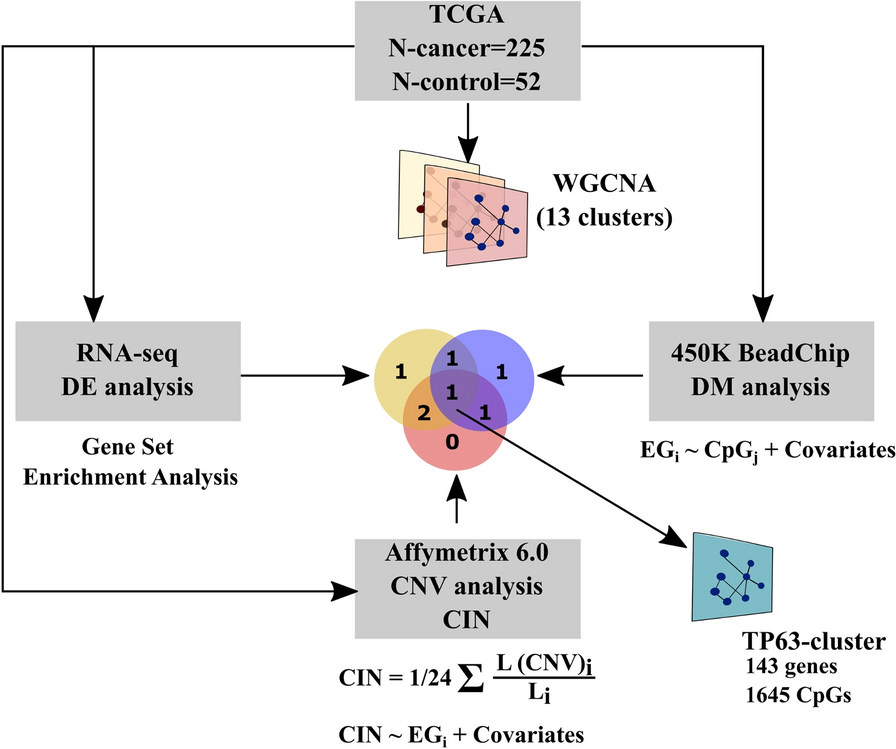
To avoid interference with neurodevelopmental processes, we performed tamoxifen (Tam) induction of Camk2a-NuTRAP in mature adult mice at 3 months of age (3mo). This timing was chosen to circumvent deficits in spatial learning, contextual fear memory, and presynaptic structure that can arise after perturbing Camk2a expression during early neurodevelopment [42, 43]. Brains were collected one month following Tam induction and sectioned sagittally for immunohistochemical analysis.
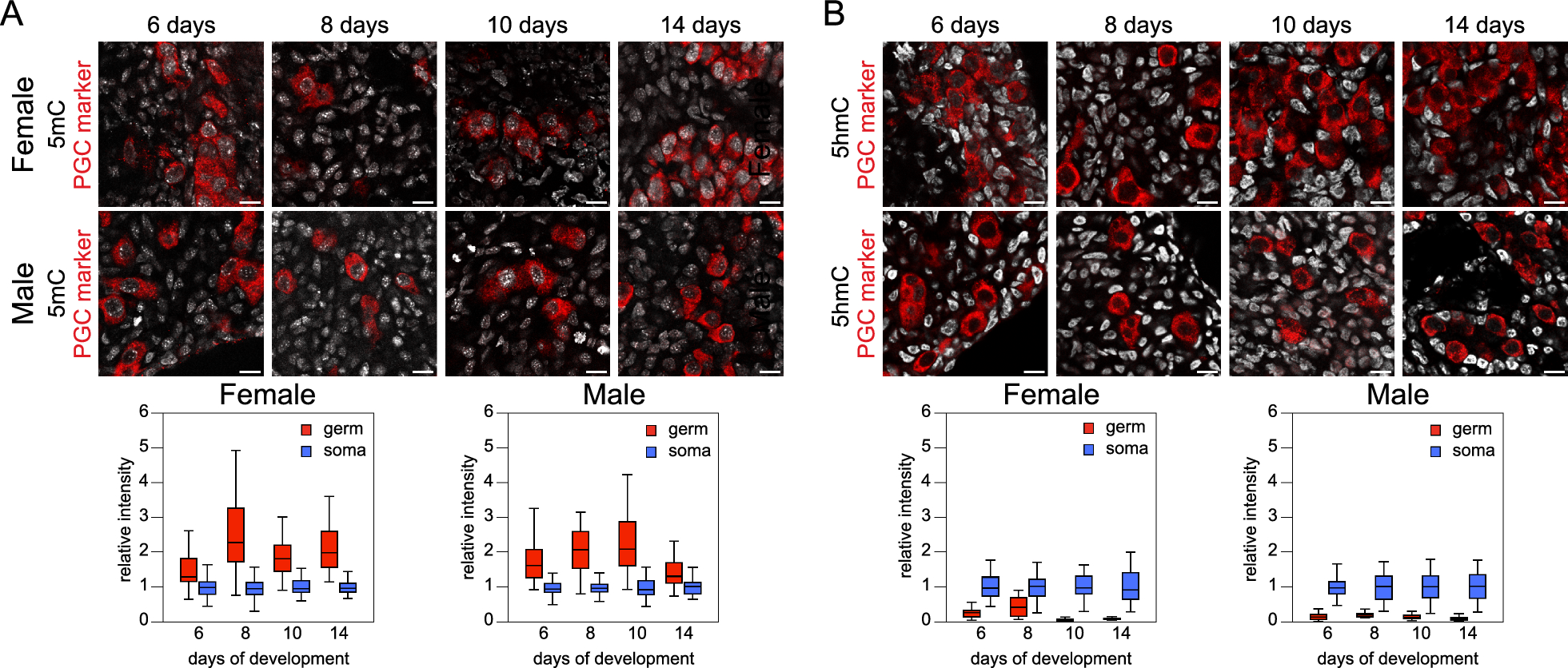
Immunostaining of Camk2a-NuTRAP (Camk2a-cre/ERT2+; NuTRAP+) brains showed EGFP and mCherry colocalization in cells expressing the pan-neuronal marker NeuN. No EGFP or mCherry expression was seen in Camk2a-cre negative counterparts (Fig. 1A) and minimal expression was observed in a portion of NeuN+ cells of Camk2a-NuTRAP (-Tam) brains, which is consistent with previous reports (Additional file 10: Figure S1A-B) [36, 44]. Camk2a-NuTRAP (+ Tam) brains show no expression of EGFP in microglial (CD11b+), endothelial (CD31+), or astrocytic (GFAP+) cells (Additional file 10: Figure S1C–E). Collectively, these findings indicate a robust neuronal-specific and tamoxifen-dependent induction of the NuTRAP allele. This validation ensures that the experimental manipulations specifically target neuronal cells while minimizing any confounding effects on other cell types in the brain.
Fig. 1
Validation of neuronal translatome enrichment in TRAP-RNA from Camk2a-NuTRAP mouse hippocampus. A Imaging of the hippocampal dentate gyrus demonstrated EGFP and mCherry co-expression in NeuN + cells. B TRAP-isolated hippocampal RNA from input, negative, and positive fractions were assessed by qPCR for enrichment and depletion of canonical marker genes for microglia, astrocytes, oligodendrocytes, and neurons. Mean relative gene expression ± SEM scaled to input for each gene. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by RM one-way ANOVA with Tukey’s multiple comparison test across fractions (n = 4/group). C RNA-seq was performed for all fractions (n = 4/group). Principal component analysis shows separation of the positive from input and negative fraction samples in the first component. D Cell-type marker gene lists were examined for fold change (Positive/Input) enrichment or depletion shows enrichment of neuronal markers and depletion of other cell-type markers in the positive fraction. E CIBERSORTx calculation of cell type composition of each fraction. The positive fraction is estimated to contain 100% neurons. F Genes with significant enrichment (2111) or depletion (2897) in the positive compared to input fraction were identified (FC >|1.25|, p < 0.05, Benjamini Hochberg multiple testing corrections). G–J Gene Ontology enrichment analysis and Ingenuity Pathway Analysis performed on significantly enriched or depleted genes (Positive/Input fraction) identified in E
Validation of neuronal translatome enrichment from TRAP-isolated RNATranslating RNA was isolated from the hippocampus of Camk2a-NuTRAP mice via the TRAP method (Translating Ribosome Affinity Purification) [41]. Subsequent RT-qPCR of RNA from the input, negative, and positive TRAP fractions showed a significant enrichment of neuronal marker genes (Camk2a, Hpca, Caln1, Kcnip4, Stmn2, and Snap25) in the positive fraction compared to input and negative fraction. Conversely, there was a depletion of microglial (Cx3cr1 and Itgam), astrocytic (Aldhl1l and Gfap), and oligodendrocytic (Mog) marker genes in the positive fraction as compared to the input and negative fraction (Fig. 1B; Additional file 1B).
To further characterize the neuronal translatomic profile, TRAP-isolated RNA was subjected to RNA-Seq. Principal Component Analysis (PCA) of all expressed genes revealed clear separation of the positive fraction from input and negative fraction in the first component (Fig. 1C). To validate the TRAP-enrichment of neuronal genes and depletion of other cell type-specific genes, we used marker lists generated from previous cell sorting studies [41] (Additional file 2). Enrichment of neuronal genes and depletion of astrocytic, microglial, oligodendrocytic, and endothelial genes was evident in the positive fraction as compared to input (Fig. 1D; Additional file 3A). Notably, there was a high fold depletion of markers for minority cell types like glia and smaller fold-change enrichment for neurons, which make up the majority of the input.
To estimate the cell type composition of the input, negative and positive fractions, we employed CIBERSORTx [45] using established cell type marker lists [41]. This analysis revealed that the input contained the expected variety of cell types at the expected proportions (astrocytes, microglia, neurons, oligodendrocytes, and endothelial cells). The negative fraction demonstrated a depletion of neuronal cells, whereas the positive fraction was estimated to be entirely represented by neurons (~ 100%) (Fig. 1E; Additional file 3B).
Ingenuity Pathway Analysis and Gene Ontology analysis of the significantly enriched and depleted genes in the positive fraction vs input (Fig. 1F; Additional file 3C,D) revealed enriched genes regulating excitatory neuronal biological processes and functions such as those involved in synaptic structure, maintenance and plasticity (Fig. 1G, H; Additional file 3E,F). On the other hand, depleted genes were involved in lipid metabolism, immune response, and vascular formation and maintenance, indicating a depletion of genes involved in non-neuronal pathways (Fig. 1I, J; Additional file 3G, H). Moreover, the positive fraction enrichment of genes involved in spatial learning, a major function of hippocampal neurons, further demonstrated the specificity and relevance of the model in capturing neuronal-specific transcripts [46, 47]. These findings provide valuable insights into the enriched and depleted gene sets within the neuronal translatome, shedding light on the functional processes and pathways associated with neuronal identity and function of the hippocampus.
Validation of neuronal gDNA isolation from INTACT Whole Genome Bisulfite SequencingTo ensure the purity of the positive fraction obtained through INTACT isolation (Isolation of Nuclei in TAgged in specific Cell Types), expression of EGFP within the nucleus [32] was assessed by confocal microscopy. EGFP-positive nuclei surrounded by streptavidin beads were observed in the positive fraction (Fig. 2A). In contrast, the input showed a mixture of EGFP-positive and EGFP-negative nuclei (Fig. 2B), while the negative fraction exhibited no EGFP expression (Fig. 2C).
Fig. 2
Validation of neuronal genome enrichment in Camk2a-NuTRAP mouse hippocampus by INTACT-BS seq. A–C Confocal fluorescent microscopy images from positive, input, and negative INTACT nuclei isolations. D INTACT-isolated gDNA from the hippocampus of Camk2a-NuTRAP mice was bisulfite converted and whole genome levels of CG modifications measured for input, negative, and positive fractions. CG modifications from previously published neuronal methylation studies utilizing various brain regions (hippocampus and cortex) and isolation techniques (Camk2a INTACT, NeuN+ sorting, and single cell) were compared to Camk2a-NuTRAP CG modifications. E Whole genome CH modifications were measured for input, negative, and positive fractions. CH modifications from the same neuronal methylation studies from D were compared to Camk2a-NuTRAP CH modifications (**p < 0.01 by one-way ANOVA with Tukey’s multiple testing correction)
To assess DNA modifications in the positive fraction, whole genome oxidative bisulfite sequencing (WGoxBS) was performed on INTACT-isolated gDNA from the input, negative, and positive fractions to measure mC and hmC in the CG and CH contexts. First, the bisulfite-only arm, which detects a combined signal of mC and hmC (total modifications), was compared to previously published neuronal bisulfite sequencing data. Total CG and CH modification levels from the positive fraction were similar to previously published neuronal bisulfite sequencing modification studies [2, 48,49,50] (Fig. 2D–E). These findings provide evidence for the isolation of neuronal-specific genomic DNA, supporting the validity of the INTACT isolation method and the subsequent analysis of DNA modifications in the positive fraction.
Neuronal epigenome analysis using whole genome oxidative bisulfite sequencingTo distinguish between mC and hmC, the oxidative bisulfite sequencing (oxBS-Seq, mC only) arm was subtracted from the bisulfite sequencing (BS-Seq, mC + hmC) arm for INTACT-isolated DNA from the input, negative, and positive fractions (Additional file 10: Figure S2A). Conversion efficiency, measured by spike-in controls, was close to 100% with no significant variance between samples or groups (Additional file 10: Figure S2B–C, Additional file 4). Comparing the different fractions, the positive fraction exhibited significantly lower mCG and higher levels of hmCG and mCH when compared to input and negative fractions (Fig. 3A–C). Non-CG hydroxymethylation (hmCH) was detected at low levels near background (< 1%) and was not significantly different between fractions (Fig. 3D).
Fig. 3
Profile of hippocampal neuronal DNA modifications by whole genome oxBS-seq. INTACT hippocampal gDNA from input, negative and positive fractions was taken for whole genome bisulfite and oxidative bisulfite sequencing. A–D Total genomic levels of mCG, hmCG, mCH, and hmCH (n = 4/group; one-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001). Levels of mCG were lower and hmCG and mCH were higher in the positive fraction. E–G mCG, hmCG, and mCH averaged over 200 nucleotide bins from 4 kb upstream, within the gene body, and 4 kb downstream of neuronal marker genes in the positive fraction and input. H–J Average mCG, hmCG, and mCH for positive fraction and input 4 kb upstream of the TSS, within the gene body, and 4 kb downstream of the TES of neuronal genes revealed lower mCG and higher hmCG and mCH in gene bodies and downstream (n = 4/group; paired two-tailed t-test between input and positive fraction, *p < 0.05, **p < 0.01, ***p < 0.001)
Deeper sequencing (1-2X) was performed on the input and positive fraction (Additional file 5), with the knowledge that only 0.001X coverage is required for obtaining accurate whole genome and repeat element modification levels within 1% [51]. Distribution of DNA modifications (mCG, hmCG, and mCH) was also mapped across genic regions (Promoter, Gene Body, Downstream) of neuronal marker genes (Additional file 2). In the positive fraction, the intragenic/gene body and downstream regions of neuronal marker genes showed significantly lower mCG levels and significantly higher mCH and hmCG levels compared to input (Fig. 3E–J). Neuronal marker genes had lower mCG, hmCG, and mCH at the TSS compared to all genes (Additional file 10: Figure S3), with lower mCG observed across the gene body of neuronal genes compared to all genes (Additional file 10: Figure S3A).
Comparison of DNA modifications across three CNS cell typesWe previously validated the use of the NuTRAP construct in two mouse lines for isolation of gDNA and RNA from astrocytes and microglia [41]. To compare the DNA modification profiles between neurons, astrocytes and microglia, previously published WGoxBS sequencing data from the positive fractions of Aldh1l1-NuTRAP and Cx3cr1-NuTRAP (GSE140271) were compared to Camk2a-NuTRAP (present study, GSE228044).
Whole genome total CG modifications (by BS-Seq) were consistent across cell types, as well as between INTACT WGoxBS and single nuclei methylome studies (snmC) [49, 52] (Fig. 4A). Alternatively, whole genome total CH modifications were consistent within cell types, with neuronal levels being nearly twice as high as astrocytes or microglia (Fig. 4B). When distinguishing between mCG and hmCG, neurons exhibited higher hmCG levels and lower mCG levels compared to astrocytes and microglia (Fig. 4D–E). To better understand the origin of observed DNA modification differences between cell types, we assessed the cell type-specific expression of modification regulators [DNA methyltransferases (DNMTs), Ten–eleven translocases (TETs), and thymine DNA glycosylase (TDG)]. Microglia had significantly higher DNMT (Dnmt1, Dnmt3a, and Dnmt3b) expression than astrocytes and microglia, aligning with microglia having the highest levels of mCG among the three cell types (Fig. 4C). Surprisingly, despite having the lowest hmCG of the three cell types assessed, microglia also express TETs (Tet1, Tet2, and Tet3) at a significantly higher level compared to neurons and astrocytes (Fig. 4C). TET2 has been previously shown to regulate the microglial type I interferon-mediated inflammatory response upon LPS administration [53], pointing to a potentially dynamic role for microglial hydroxymethylation in modulating cell phenotype.
Fig. 4
Comparison of DNA modifications across three CNS cell types. Whole genome total CG modifications A and CH modifications B from INTACT-isolated gDNA from neurons (hippocampus), astrocytes (half brain), and microglia (half brain) were compared to two single nuclei methylome studies [49, 52]. C TRAP RNA-seq expression of DNA modification regulators in neurons, astrocytes, and microglia (n = 3–6/group; one-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) data presented at reads per kilobase mapped. D–F Whole genome, repetitive element, and non-repetitive element mCG, hmCG, and mCH levels for neurons, astrocytes, and microglia. (n = 4/group; two-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
On the other hand, TDG, which mediates base-excision repair in active demethylation and single-strand break repair, was most highly expressed in neurons (Fig. 4C). As such, the methylation and demethylation cycle may serve as a source of site-specific neuronal single-strand breaks that have been previously observed within enhancer elements [54]. DNMTs, TETs, and TDG were also examined in public single cell gene expression repositories (Tabula Muris [55], Allen Brain [56], and Aging Mouse Brain atlases (young timepoint only) [57]), but due to comparative insensitivity of scRNA-Seq to TRAP-Seq, many of these genes were at the limit of detection and did not demonstrate clear patterns of differences between cell types (Additional file 10: Figure S4).
Repetitive elements comprise over 50% of the genome and are thought to play an important role in neuronal differentiation and maturation [58, 59]. To determine the genomic localization of the observed cellular DNA modification differences, the levels of DNA modifications in whole genome, repeat elements, and non-repeat elements were assessed in neurons, astrocytes, and microglia. mCG levels were lower in neurons across the whole genome, repeat, and non-repeat elements compared to astrocytes and microglia (Fig. 4D). Conversely, hmCG levels were significantly higher in neurons across repeat and non-repeat elements than astrocytes and microglia, with microglia exhibiting the lowest hmCG levels among the three cell types (Fig. 4E). In the CH context, a similar pattern was observed across the genome and when split between repeat and non-repeat elements (Fig. 4F).
Furthermore, when examining the split of whole genome levels into repeat and non-repeat elements, it was observed that repetitive elements had significantly higher mCG levels, while non-repetitive elements had significantly lower mCG levels compared to whole genome levels (Fig. 4D). Conversely, there was significantly lower hmCG levels in repetitive elements compared to the whole genome levels in neurons and astrocytes, with no difference between non-repetitive elements and whole genome levels for neurons, astrocytes or microglia (Fig. 4E).
In general, CG modification levels between cell types of repetitive and non-repetitive elements followed the pattern observed in whole genome levels. On the other hand, mCH levels were consistently the highest in neurons followed by astrocytes and then microglia, regardless of the genomic context across whole genome, non-repetitive, or repetitive elements (Fig. 4F). Unlike CG modifications, mCH levels were observed to be nearly identical across repetitive and non-repetitive elements. These findings provide insights into the cell type-specific distribution of DNA modifications across different genomic regions, including the impact of repeat elements, and highlight the distinct epigenetic landscapes in neurons, astrocytes, and microglia.
To further validate the whole genome CG methylation and hydroxymethylation values obtained by WGoxBS, long-read nanopore sequencing and native CG methylation and hydroxymethylation calling was performed on separate INTACT-isolated high molecular weight gDNA from neurons, astrocytes, and microglia (n = 2/group; PRJNA1026932). As was observed from WGoxBS, neurons had lower mCG (Fig. 5A) and higher hmCG (Fig. 5B) levels compared to astrocytes and microglia. The absolute values obtained were also highly consistent with the WGoxBS data and between biological replicates (Fig. 5C). The pattern of modification levels between cell types is observed across entire chromosomes, as is represented by Chromosome 15 (Fig. 5C). Overall, the same pattern of cell type differences in mCG and hmCG were observed with both WGoxBS and nanopore sequencing, offering further validation for the DNA modification levels reported above. A table of average whole genome mCG and hmCG measured with WGoxBS, conversion corrected oxBS, and Nanopore can be found in Additional file 10: Table S1.
Fig. 5
Native detection of DNA modifications with nanopore long-read sequencing. Nanopore long-read sequencing was performed on INTACT-isolated high molecular weight gDNA from neurons, astrocytes, and microglia (n = 2/group). Native mCG and hmCG calling was performed to obtain total whole genome %mCG A and %hmCG B. %modC (%mCG or %hmCG) was plotted across chromosome 15 C to demonstrate modification differences between cell types and reproducibility across biological replicates. Modification values were smoothed in CpG-only coordinate space (One-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.01)
Repetitive elements are a known source of somatic mosaicism in the brain, and their aberrant activity (particularly LINE1) is implicated in several neurological and neurodegenerative diseases [60]. We next compared the DNA modification levels between neurons, astrocytes, and microglia in specific repeat elements: long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long terminal repeats (LTRs), and simple repeats. Neurons had lower mCG levels compared to astrocytes and microglia in all analyzed repeat elements (LINEs, SINEs, LTRs, and simple repeats) (Fig. 6A). Consistent with the whole genome levels, neurons had the highest level of hmCG and mCH within LINEs, SINEs, LTRs, and simple repeats, whereas microglia had the lowest levels of these modifications (Fig. 6B–C). Compared to whole genome levels, mCG levels were higher within repetitive elements (LINEs, SINEs, LTRs, and simple repeats), whereas repetitive hmCG and mCH levels were lower than whole genome. The only exception to this was mCH levels within simple repeats [2.51% (neuron), 1.54% (astrocyte), 1.17% (microglia)], which were higher than whole genome levels [1.46% (neuron), 0.85% (astrocyte), 0.67% (microglia)] (Fig. 6A–C). Additionally, simple repeats use more mCH and less mCG compared to other specific repeat elements analyzed (Fig. 6A, C). The mCG and hmCG levels in LINEs, SINEs, and LTRs were also assessed from nanopore long-read sequencing, and were overall consistent with WGoxBS (Additional file 10: Figure S5). Generally, the modification patterns between specific repeat elements followed the patterns observed at the whole genome level. However, there were differences in the absolute levels of DNA modifications depending on the specific repeat element analyzed.
Fig. 6
Repeat element DNA modifications in the CNS. mCG A, hmCG B, and mCH C levels of LINE, SINE, LTR, and Simple Repeat elements for neurons, astrocytes, and microglia. (n = 4/group; one-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
Differential DNA modifications are enriched at cell-type specific transcription factor motifsTo determine the genomic localization of DNA modification differences between cell types, pairwise differentially modified regions (DMRs) were identified for each DNA modification type. Differential mCG regions (DMCGRs) consisted of both hyper- and hypo-methylation (Fig. 7A) and were primarily comparison-specific, with the greatest overlap being between glia (microglia and astrocytes) and neurons (Fig. 7B,C). DMCGRs were distributed across the genome for each comparison and ranged from –100 to 100% difference (Fig. 7D–F).
Fig. 7
Differentially methylated CG regions. Differentially methylated CG regions (DMCGRs) were determined between cell types. Distribution of mCG differences A was plotted, along with overlap of hyper- B and hypo- C DMCGRs between the three comparisons. Genomic distribution and magnitude of DMCGRs for astrocytes vs neurons D, microglia vs neurons E, and astrocytes vs microglia F. Relative over- and under-representation in genic features for astrocytes vs neurons G, microglia vs neurons H, and astrocytes vs microglia I. Top enriched transcription factor binding motifs for astrocytes vs neurons J, microglia vs neurons K, and astrocytes vs microglia L. Genic regions containing no DMCGRs were notated as “n.d.” (Woolf logit method for 95% confidence intervals, Fisher’s exact test for two-sided p-values; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
To analyze the localization of DMCGRs within genic contexts, over- and under-representation analysis was performed (as compared to random distribution across the background). DMCGRs for all three comparisons were over-represented in gene body regions, distal promoters, and intergenic contexts, while being under-represented in proximal promoters (Fig. 7G–I). Despite being over-represented within most cell types, astrocytic hypermethylation within distal intergenic regions compared to microglia was under-represented, demonstrating a different epigenomic patterning between individual glial cell types than between glia and neurons.
HOMER analysis was performed on DMCGRs to identify enriched motifs for each comparison (Additional file 6) [61], which revealed transcription factor binding motifs associated with cell type-specific functions. For instance, highly methylated astrocytic regions were enriched for HIF1b binding sites, while highly methylated neuron regions are enriched for Zic binding sites (Fig. 7J). These transcription factors, along with their targeted genes Npas4 and Apoe, have known roles in excitatory-inhibitory balance in the central nervous system [62, 63], and lipid transport in cerebellar astrocytes [64, 65], respectively. As previously mentioned, mCG hypermethylation is generally associated with transcriptional repression. Thus, hypermethylation of these essential transcription factors have downstream implications for cell type-specific functions of CNS cells.
In the comparison between microglia and neurons (Fig. 7K), hypermethylated microglial regions were found to be enriched in Lhx2 binding sites, which inhibit Gfap expression and promote neurogenesis in the hippocampus [66]. Hypermethylated neuronal regions were enriched in Fli1 binding sites, which are implicated in the shift from homeostatic to ramified microglia through Spi1 and Runx1 [67,68,69]. In the comparison between astrocytic and microglial DMCGRs (Fig. 7L), hypermethylated astrocytic regions were enriched in NF1-halfsite binding sites, which has downstream regulators such as Sp1, Mef2c, and Sall1, all essential modulators of homeostatic microglia [70]. Microglial hypermethylation was enriched in PU.1 binding sites, and although is most well-known for its function as a master regulator of microglia, regulates the astrocytic maturation marker Runx2 during development as well [69]. Together, motifs in DMCGRs followed the expected inverse relationship with binding sites of known cell identity-related transcription factors.
Differential hydroxymethylated CG regions (DhMCGRs) between cell types consisted of both hyper- and hypo-hydroxymethylation (Fig. 8A), and were mainly comparison-specific. The greatest overlap was in hypo-hydroxymethylated regions between glial cells and neurons (Fig. 8B, C), and DhMCGRs were distributed throughout the genome for all three comparisons (Fig. 8D–F). Analysis of the genomic distribution of DhMCGRs revealed over-representation in genic regions and distal promoter regions, with under-representation in proximal promoter regions across all comparisons (Fig. 8G–I). Specifically, neuronal and astrocytic hyper hydroxymethylation was under-represented in distal intergenic regions, whereas over-representation was observed in microglia (Fig. 8G–I).
留言 (0)