
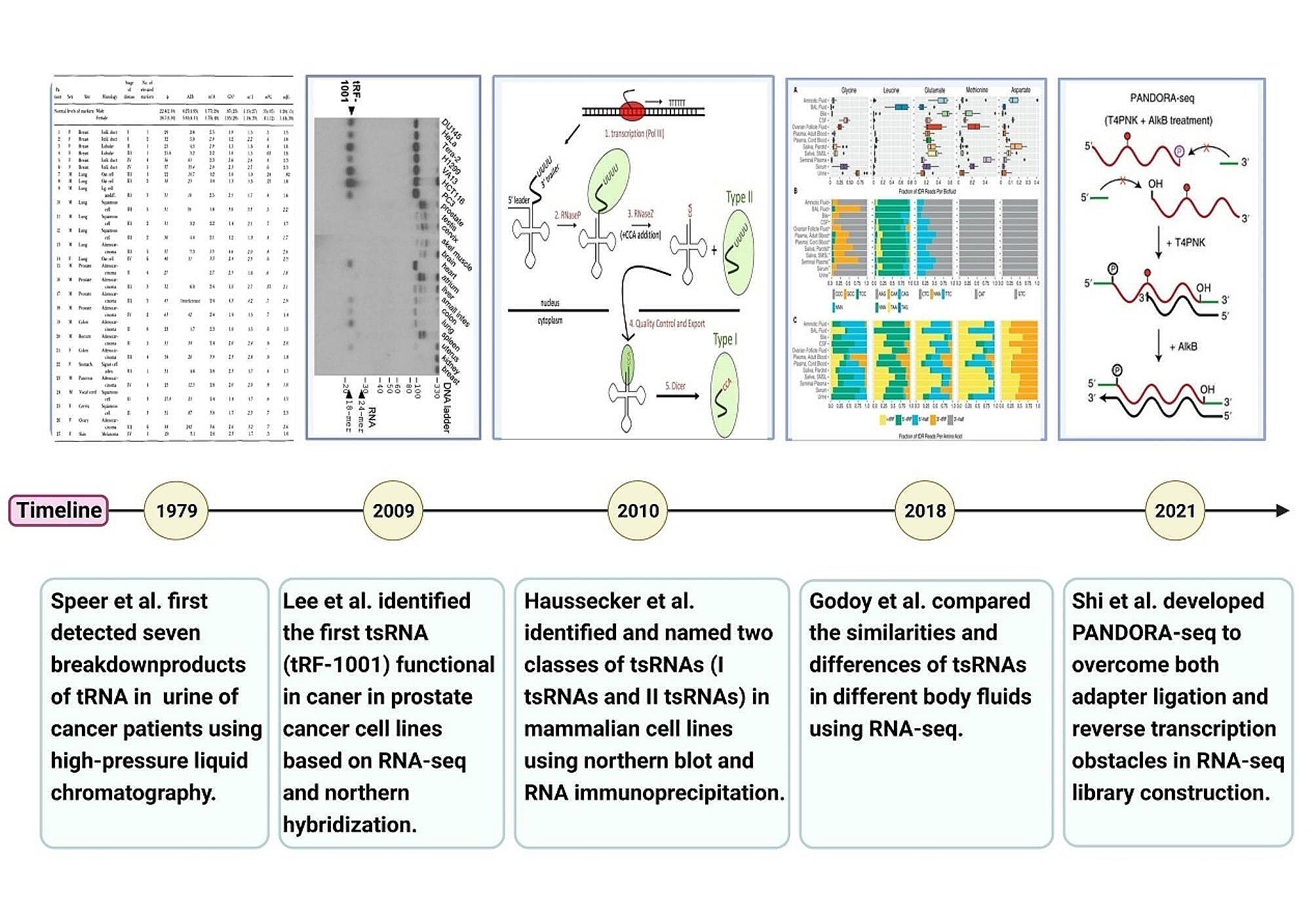
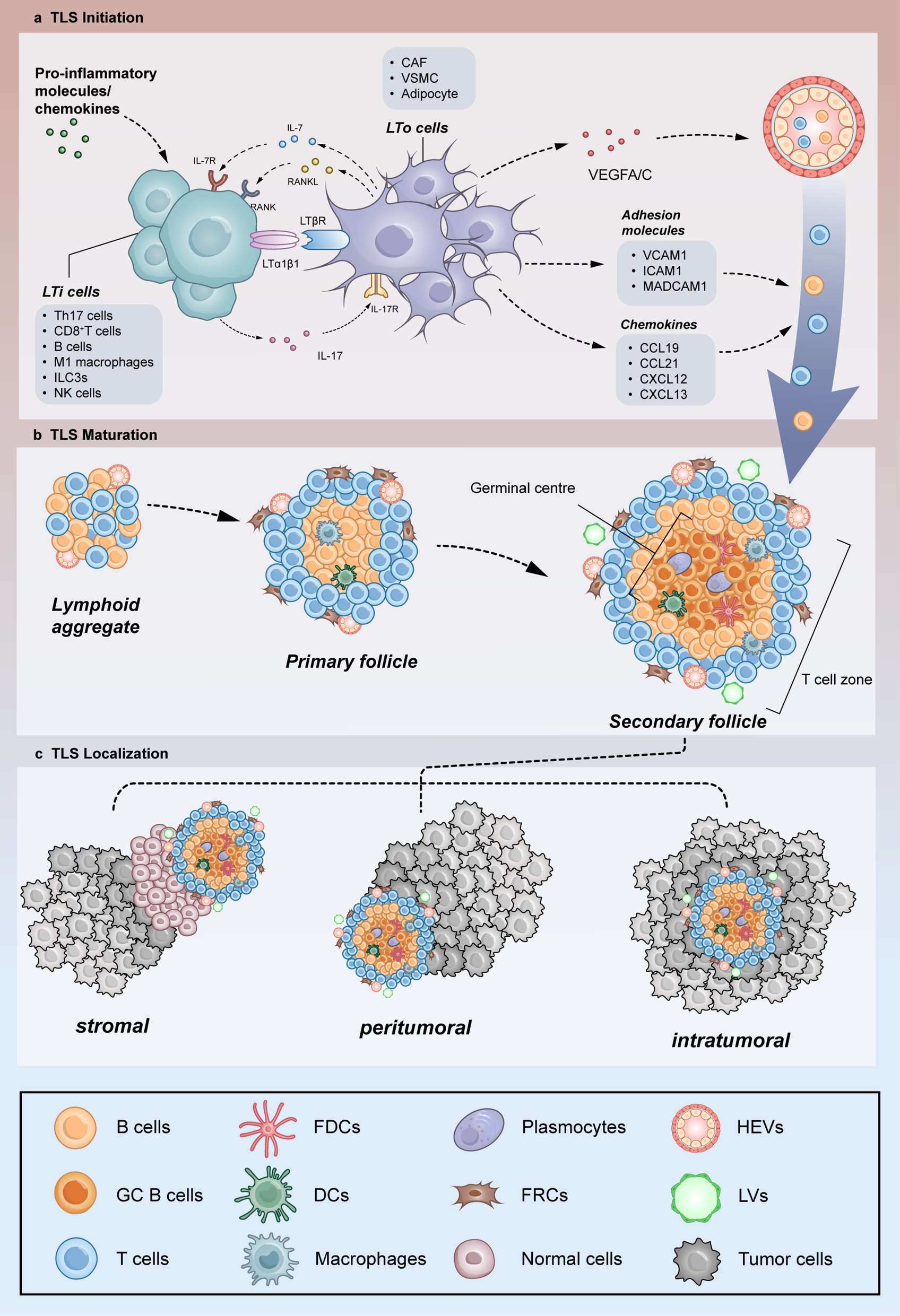
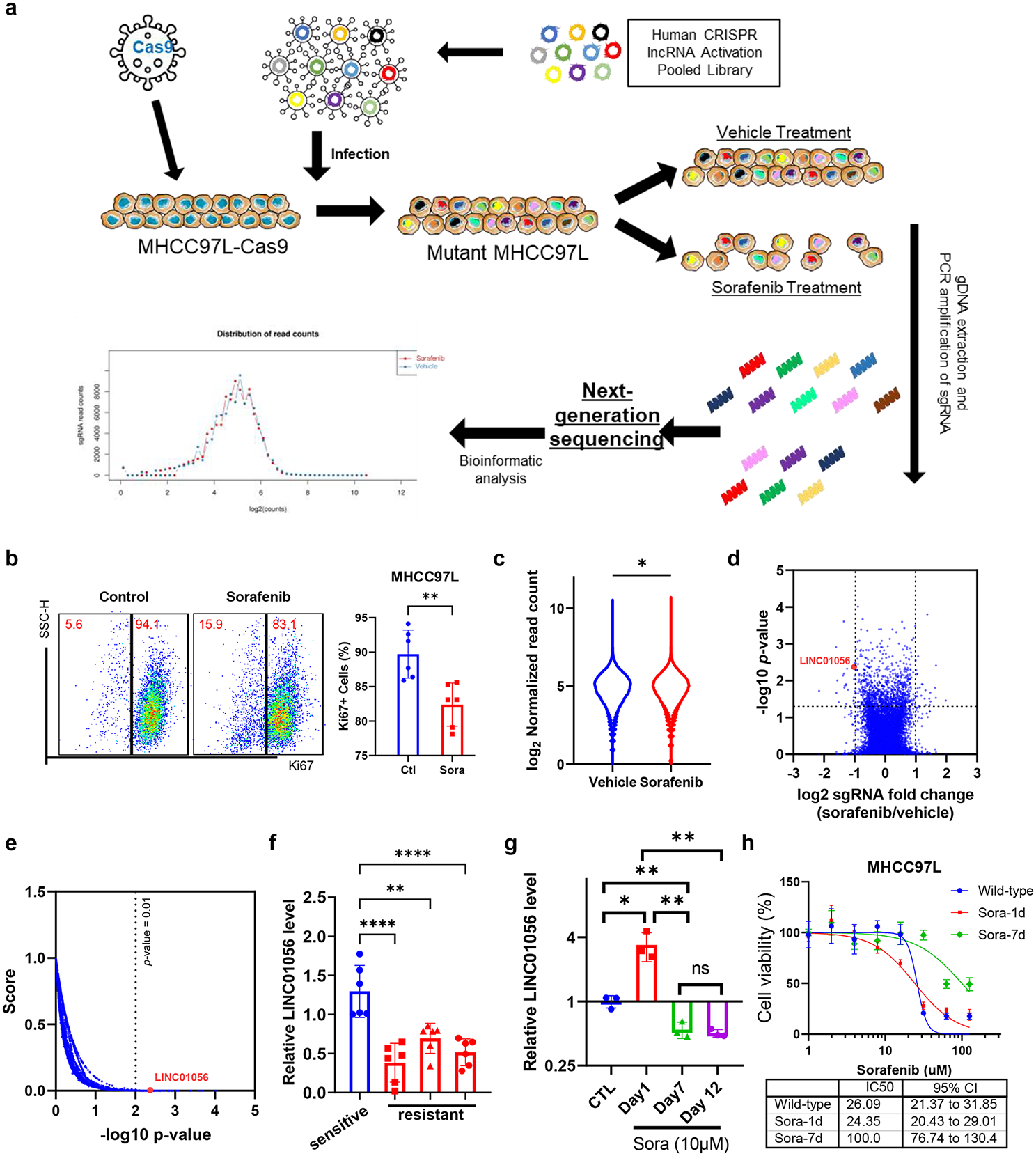
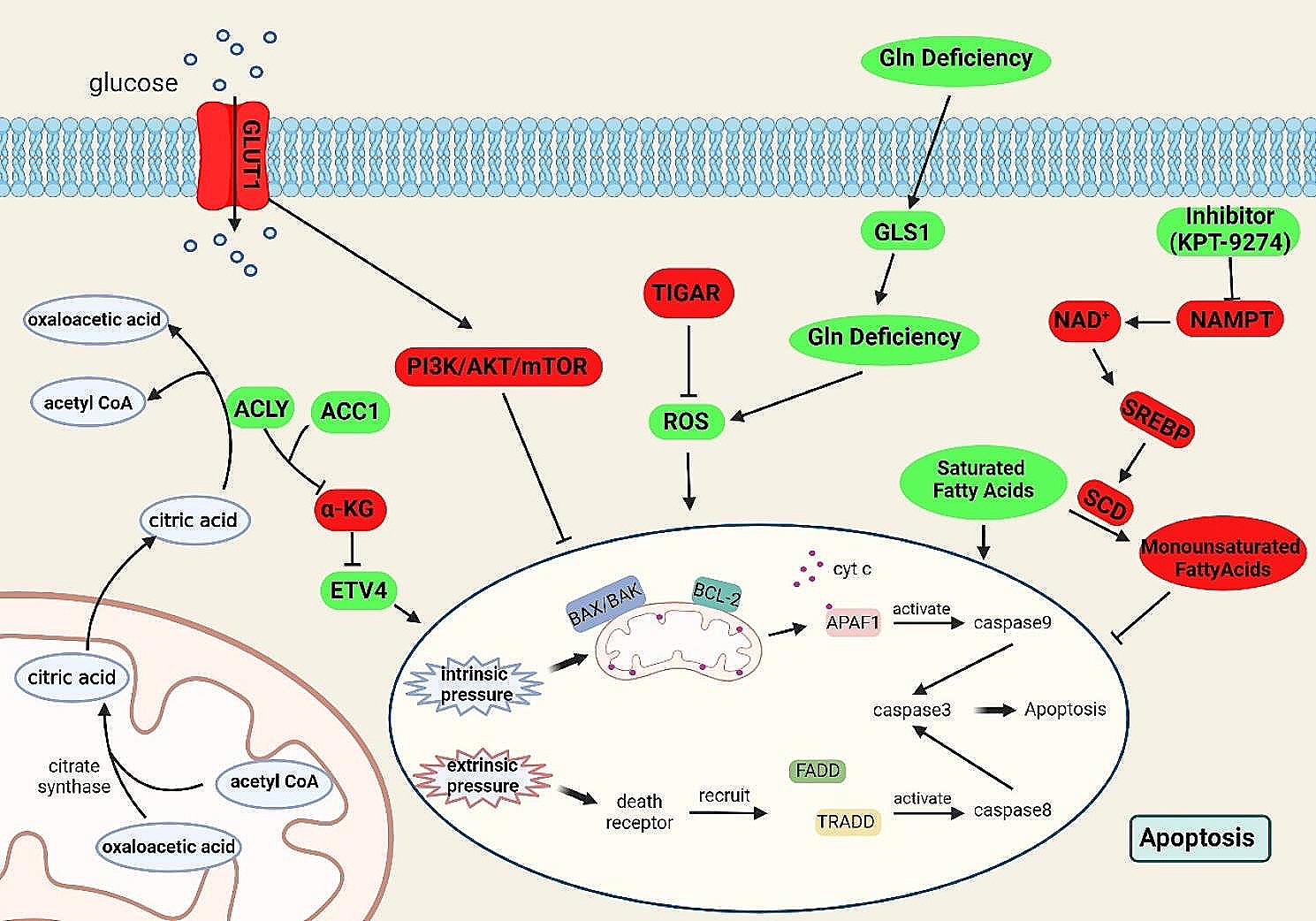
記住我


To investigate the clinical significance of EZH2 and H3K27me3 in OC, we examined the protein level of EZH2 and the level of H3K27me3 in a cohort consisting of 105 OC patients using an IHC approach. The IHC data indicated that the EZH2 and H3K27me3 levels were not significantly correlated in our cohort of OC patients (Fig. 1A). Patients with high levels of EZH2 exhibited a worse prognosis than those with low levels of EZH2, whereas high levels of H3K27me3 predicted a better prognosis in OC patients (Fig. 1B and C). Based on the patterns of EZH2 expression and the H3K27me3 level, we classified OC patients into four subgroups: EHZ2high/H3K27me3high (11%), EHZ2low/H3K27me3low (45%), EHZ2low/H3K27me3high (19%) and EHZ2high/H3K27me3low (25%) (Fig. 1D). In addition, after stratification of patients by H3K27me3 levels, the EZH2low/H3K27me3high pattern correlated with the better survival outcomes, and patients in the EZH2high/H3K27me3high subgroup still had a worse prognosis (Fig. 1E). These observations are consistent with a previous study showing that overexpression of EZH2 with distinct level patterns of H3K27me3 levels correlates with different prognoses in patients with OC, suggesting a decoupling of EZH2 and H3K27me3 in OC tumorigenesis [21]. Furthermore, we showed that in both The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) projects [22] and in our own in-house data, EZH2 was overexpressed in OC tissues compared to the normal tissues, whereas the H3K27me3 level was decreased in OC tissues compared to normal fallopian tube tissues (Fig. 1F-H). Together, these results suggest that the oncogenic role of EZH2 may be independently of its catalytic activity in OC.
Fig. 1
Overexpression of EZH2 with low H3K27me3 correlates with the worst prognosis in patients with OC
(A) Correlation analysis between EZH2 and H3K27me3 levels in the OC cohort (n = 105). P value was determined by Pearson’s correlation test
(B) Kaplan-Meier survival analysis for the OC cohort (n = 105), which was divided into EZH2high and EZH2low subgroups based on the cutoff (cutoff = 6) calculated by the X-Tile software. The P value was calculated by log-rank test. *P < 0.05
(C) Kaplan-Meier survival analysis for the OC cohort (n = 105), which was divided into H3K27me3high and H3K27me3low subgroups based on the cutoff (cutoff = 6) calculated by the X-Tile software. The P value was calculated by log-rank test. *P < 0.05
(D) Representative IHC images of EZH2 and H3K27me3. EZH2 and H3K27me3 levels were detected by IHC staining in a cohort of 105 patients with OC. Scale bar: 100 μm
(E) Kaplan-Meier survival analysis for OC samples (n = 105) in four subgroups, which was classified based on EZH2 (cutoff = 6) and H3K27me3 (cutoff = 6) levels. The P value was calculated by log-rank test. *P < 0.05, **P < 0.01
(F) EZH2 log2(TPM) value in normal ovarian tissues (GTEx, n = 88) and ovarian cancer tissues (TCGA, n = 426). Data are presented as the mean ± SEM. *P < 0.05
(G) Staining score of EZH2 and H3K27me3 levels in fallopian tube and OC tissues. IHC score of EZH2 and H3K27me3 was determined as the scores for the proportion of positively stained tumor cells (1, 1–25%; 2, 26–50%; 3, 51–75%; 4, > 75%) and staining intensity (0, no staining; 1, weak; 2, moderate; 3, strong) by each investigator were multiplied and then averaged. *P < 0.05, ***P < 0.0001
(H) Representative images of EZH2 and H3K27me3 staining in fallopian tube (n = 33) and OC (n = 105) tissues. Scale bar: 100 μm
EZH2 functions as an oncogenic driver to promote OC growth independently of its catalytic activityTo investigate the function of EZH2 in OC, we applied 4 inhibitors of EZH2 in three OC cell lines that show high expression of EZH2. The first EZH2 inhibitor described, DZNep has been reported to induce EZH2 degradation and inhibit histone methylation [23]. YM281 was recently reported as a small molecule EZH2 degrader [24]. GSK126 and EPZ-6438 are catalytic inhibitors of EZH2 that bind to its SET domain and compete with the cofactor S-adenosylmethionine (SAM) without affecting the protein stability of EZH2 [20]. Intriguingly, in both the clonogenic growth and cell proliferation assays, DZNep and YM281 but not GSK126 or EPZ-6438 suppressed the growth of OC cell lines (Fig. 2A and B, Supplementary Figure S1A). Using immunoblotting assay, we observed that both DZNep and YM281 could reduce the levels of EZH2 and H3K27me3, whereas GSK126 and EPZ-6438 could decrease only the level of H3K27me3 and did not affect the EZH2 protein level (Fig. 2C). These results suggest that the oncogenic function of EZH2 in OC is independent of its catalytic activity. Consistent with these findings, we observed that DZNep and YM281 but not GSK126 attenuated the capacity of tumor sphere formation (Fig. 2D and Supplementary Figure S1B). The differential efficacies of DZNep, YM281 and EPZ-6438 were further demonstrated in a xenograft model. After continuous dosing with DZNep and EPZ-6438, we found that DZNep but not EPZ-6438 significantly suppressed tumor growth in vivo (Fig. 2E-F). In DZNep-treated tumors, the Ki67 level was decreased concomitantly with the EZH2 but not the H3K27me3 level, further supporting the idea that the oncogenic role of EZH2 is independent of its catalytic methylation activity (Fig. 2G and Supplementary Figure S1C). These results revealed that a reduction in the level of EZH2 but not inhibition of its catalytic activity suppresses the progression of OC.
Fig. 2
EZH2 functions as an oncogenic driver to promote OC growth independently of its catalytic activity
(A) Colony formation assay in three OC cell lines. Cells were treated with DMSO or four different EZH2 inhibitors with indicated concentrations for 12 days. Growth medium was changed and inhibitors were also replenished every 3 days. Images are representative of three independent experiments
(B) Cell growth curve of 3 OC cell lines treated with DZNep (1µM), YM281 (5µM), GSK126 (5µM) or EPZ-6438 (5µM) for 96 h. Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
(C) Immunoblotting analysis of EZH2 and H3K27me3 in 3 OC cell lines treated with four different EZH2 inhibitors with indicated concentrations
(D) Tumorsphere formation assay of OC cells treated with DZNep (1µM), YM281 (5µM), GSK126 (5µM) for 10–12 days. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001
(E-F) Xenograft tumor growth (E) and tumor weight (F) of OVCAR8 cells in nude mice treated with saline as control, EPZ-6438 at 200 mg/kg intragastrically daily, or DZNep at 1 mg/kg intraperitoneally twice a week. n = 7 per group. Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001(Two-way ANOVA with Tukey’s post hoc test)
(G) IHC analysis of EZH2, H3K27me3, and Ki67 levels in the excised tumors from (F). Data are presented as mean ± SEM. ***P < 0.001
(H) Immunoblot analysis of EZH2 and H3K27me3 levels in shEZH2-inducible OVCAR8 cell lines with wildtype or mutant EZH2 restoration. Expression of EZH2-WT and the EZH2-ΔSET was detected by an EZH2 antibody against residues surrounding Arg354 of human EZH2 protein (SET domain locates in 612–727 amino acids). GAPDH and Histone H3 were used as loading controls
(I) Cell growth curve of shEZH2-inducible OVCAR8 cell lines with wildtype or mutant EZH2 restoration in the presence or absence of Dox at 1 µg/mL for 96 h. Data are presented as mean ± SD. ***P < 0.001
(J) Colony formation assay in shEZH2-inducible OVCAR8 cell lines with wildtype or mutant EZH2 restoration in the presence or absence of 10ng/mL Dox for 12 days. Growth medium was changed and doxycycline was also replenished every 3 days. Images are representative of three independent experiments
(K) Tumorsphere formation assay of indicated cells treated with or without Dox at 10ng/mL for 10 days. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001 vs. EV groups
Since DZNep and YM281 suppressed the proliferation of OC cells by reducing the EZH2 level, the effect of EZH2 depletion in OC was investigated via a doxycycline-inducible Tet-On system expressing EZH2 short hairpin RNA (shRNA). The addition of doxycycline effectively induced the expression of EZH2 shRNA, which drastically decreased the level of EZH2 in three OC cell lines (Supplementary Figure S1D). EZH2 depletion not only suppressed cell proliferation but also attenuated the capacity of tumor sphere formation in OC cells (Supplementary Figure S1E and F). The oncogenic effect of EZH2 was further validated in vivo using OVCAR8 cells harboring the same system; downregulation of EZH2 expression following doxycycline treatment significantly inhibited xenograft tumor growth (Supplementary Figure S1G and H). These results provide strong evidence that EZH2 functions as a bona fide oncogene in OC.
To determine whether the catalytic activity of the SET domain is required for the oncogenic function of EZH2, we generated OVCAR8 and OVCAR5 cells stably expressing EZH2 or EZH2-ΔSET (lacking the SET domain) with the EZH2 inducible knockdown construct shEZH2#2. We introduced a silent point mutation at the EZH2#2 shRNA target site in the cloned EZH2 and EZH2-ΔSET gene sequences, which enabled us to study the differential effect of endogenous and ectopic gene expression. As expected, ectopic expression of EZH2 but not the EZH2-ΔSET restored the H3K27me3 level, which was reduced upon EZH2 silencing (Fig. 2H and Supplementary Figure S1I). However, both EZH2 and EZH2-ΔSET were able to rescue the suppressed tumor growth and tumor sphere formation induced by EZH2 silencing (Fig. 2I-K and Supplementary Figure S1J-H). Collectively, these results demonstrate that EZH2 promotes OC initiation and proliferation independently of its catalytic activity in OC.
EZH2 noncanonically occupies promoters associated with metabolic genesTo systematically investigate the gene-regulatory roles of EZH2 in OC, we carried out integrative analysis of ChIP-Seq data to profile the genome-wide occupancy of EZH2, H3K27me3 and SUZ12, a subunit in the PRC2 complex, in OVCAR8 cells. A large majority of EZH2 peaks lacked H3K27me3 and SUZ12 binding and were termed as EZH2-solo binding sites, and only a small subset of EZH2 peaks overlapped with H3K27me3 and SUZ12 peaks, specifying canonical EZH2-ensemble binding sites (Fig. 3A-C). Approximately 50% of the EZH2-solo peaks were located at promoters (Fig. 3D). To confirm that these EZH2 solo binding sites are active promoters, an additional chromatin mark H3K4me3 was analyzed. Compared to the EZH2 ensemble and H3K27me3 solo binding sites, the EZH2 solo binding sites were overwhelmingly enriched in the H3K4me3 peaks (Fig. 3E and F). These results demonstrate that EZH2 solo binding sites are prevalent in OC and potentially regulate gene activation, differing from the canonical roles of the PRC2 complex.
Fig. 3
EZH2 noncanonically occupies promoters associated with metabolic genes
(A-B) Averaged signal intensities (A) and heatmaps (B) for EZH2, H3K27me3 and SUZ12 ± 2 kb from the centres of EZH2 or H3K27me3 peaks in OVCAR8 cells
(C) Venn diagram showing the significant overlap between EZH2 and H3K27me3 binding sites identified in OVCAR8 cells
(D) Pie-chart plot showing the genomic distribution of peaks with EZH2-solo binding in OVCAR8 cells
(E-F) Averaged signal intensities (E) and heatmaps (F) for EZH2, H3K27me3, SUZ12 and H3K4me3 ± 2 kb from the centres of EZH2-solo, EZH2-ensemble or H3K27me3-solo peaks in OVCAR8 cells
(G) Scatterplot showing log2 fold change of genes expression in OVCAR8 cells after DZNep treatment or doxycycline-induced EZH2 silencing. Significant differential expression genes (DEGs) upon either treatments are indicated in blue color, upregulated DEGs in both groups are indicated in orange color, downregulated DEGs in both groups are indicated in green color. Spearman correlation between all genes is shown as a black line (R = 0.846, p < 0.0001)
(H) Venn diagram showing the significant overlap between 617 EZH2 potential targets and EZH2-solo peaks associated genes in OVCAR8 cells. Averaged signal intensities for EZH2, H3K27me3, SUZ12 and H3K4me3 ± 2 kb from the TSS of overlap genes are shown
(I) Gene set enrichment analysis for the 251 EZH2 solo targets using the KEGG and Reactome gene sets. Metabolism-related pathways are among the most enriched (left). The -log10(P-value) of metabolism-related pathways are shown (right)
(J) Metabolism-related genes are upregulated in ovarian cancer tissues, and down regulated after EZH2 reduction. A heatmap showing the relative expression of top 10 metabolic genes in (H) by calculating their Z-score in indicated RNA-Seq data
To define the EZH2 solo target genes in OC, we performed RNA-Seq analysis in OVCAR8 cells upon DZNep treatment or silencing of EZH2. The transcriptional changes occurring in response to the two different approaches of EZH2 depletion were highly correlated (Fig. 3G). DZNep treatment resulted in upregulation of 833 genes and downregulation of 850 genes, while silencing of EZH2 caused upregulation of 1007 genes and downregulation of 1127 genes (Fig. 3G). Since the noncanonical function of EZH2 is to activate gene expression, genes downregulated upon EZH2 reduction were regarded as potential EZH2 potential targets. The 617 overlapping downregulated genes were further intersected with the EZH2 solo peak genes, and the resulting 251 overlapping genes enriched in EZH2 and H3K4me3 peaks but not H3K27me3 or SUZ12 peaks were defined as EZH2 solo targets (Fig. 3H). Gene set enrichment analysis (GSEA) using the KEGG and Reactome databases revealed that the most significantly negatively enriched pathways upon EZH2 depletion were metabolic signaling pathways (Fig. 3I), suggesting a role of EZH2 in metabolic regulation in OC. Furthermore, we identified 57 EZH2 solo targets contributing to the metabolic signaling pathways overlapping between the DZNep treatment and EZH2 silencing groups. The expression profiles of these 57 metabolic genes were further verified in normal ovarian and ovarian tumor tissues from the TCGA cohort and GTEx project. Twelve of these 57 genes were upregulated (log2foldchange > 1, p-value < 0.05) in ovarian cancer (Fig. 3J). Additionally, these 12 genes were downregulated upon both pharmacological and genetic EZH2 depletion, implying that EZH2 plays its oncogenic role partially via these genes (Fig. 3J). Taken together, these results indicate that the noncanonical roles of EZH2 in OC might be associated with metabolic rewiring.
EZH2 transcriptionally upregulates IDH2 to promote metabolic rewiringOC was previously found to exhibit metabolic heterogeneity, and OXPHOS is one of the major mechanisms of energy production in OC cells [25]. Interestingly, the genes encoding isocitrate dehydrogenase 2 (IDH2) and oxoglutarate dehydrogenase L (OGDHL), which participate in oxidative phosphorylation (OXPHOS) and the tricarboxylic acid cycle (TCA cycle), were among the top 10 genes upregulated when EZH2 was inhibited (Fig. 3J and Fig. 4A). In addition, we performed gene expression profiling analysis with the RNA-Seq data from the TCGA ovarian cancer cohort. KEGG pathway analysis showed that the oxidative phosphorylation pathway was significantly enriched in ovarian cancer tissues compared to normal ovarian tissues, suggesting that the TCA cycle or oxidative phosphorylation activity plays an important role in ovarian cancer development (Supplementary Figure S2A). These results raised the possibility that EZH2 is involved in the mitochondrial TCA cycle to promote OXPHOS in OC.
Fig. 4
EZH2 transcriptionally upregulates IDH2 to promote metabolic rewiring
(A) Diagram of TCA cycle. TCA cycle related genes in Fig. 3I are indicated in orange color
(B) qRT-PCR analysis of TCA cycle related genes in OVCAR8 and OVCAR4 cells upon 5µM DZNep or GSK126 treatment for 72 h. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
(C-D) qRT-PCR analysis of EZH2 and IDH2 levels upon Dox-induced EZH2 silencing in indicated cells. IDH2 level was concomitantly reduced upon Dox-induced EZH2 silencing (C), and ectopic expression of EZH2 or EZH2-ΔSET restored the level of IDH2 (D). Data are presented as mean ± SD. *P < 0.05, **P < 0.001, ***P < 0.001
(E-F) Dual-luciferase reporter assay of IDH2 promoter activity in OC cell lines upon pharmacological or genetic inhibition of EZH2. The OVCAR8 and OVCAR4 cells were treated with 5µM DZNep or GSK126 for 48 h before luciferase reporter assay (E). The indicated cells were treated with 1 µg/mL Dox for 48 h before luciferase reporter assay (F). Data are presented as mean ± SD. **P < 0.01, ***P < 0.001
(G) ChIP assay in OVCAR8 cells. A specific antibody against EZH2 but not isotype IgG, SUZ12, or H3K27me3 could capture the fragment containing EZH2 binding site in the IDH2 promoter region, which was amplified by specific primers (top) by qRT-PCR (bottom)
(H) ChIP-seq profiling showed the ChIP-seq signal for EZH2, H3K4me3, H3K27me3 and SUZ12 at the genomic loci of IDH2 in OVCAR8 cells. ChIP-seq profiling also showed the ChIP-seq signal for EZH2 and H3K27me3 in two MLL1-rearranged leukaemia cell lines in GSE180448.
(I-J) OCR assays upon pharmacological (I) or genetic (J) inhibition of EZH2. Representative OCR pattern as a function of time (in min), normalized to total protein levels. Cells were pretreated with 5 µM DZNep, GSK126 or 1 µg/mL doxycycline for 72 h before OCR measurement. Data are shown as mean ± SD of three replicates per treatment
To determine whether EZH2 promotes TCA cycle activity, we next examined the mRNA levels of IDH2 and OGDHL upon DZNep or GSK126 treatment in OVCAR8 and OVCAR4 cells. Interestingly, IDH2 and OGDHL were downregulated upon treatment with DZNep but not GSK126, suggesting that EZH2 regulates the TCA cycle independently of its methyltransferase activity (Fig. 4B). We showed that IDH2, the rate-limiting enzyme for the TCA cycle, is significantly upregulated in OC, which prompted us to investigate the correlation between EZH2 and IDH2. We observed that the IDH2 level was concomitantly reduced upon doxycycline-induced EZH2 silencing (Fig. 4C), and that ectopic expression of EZH2 or EZH2-ΔSET restored the level of IDH2 (Fig. 4D), indicating that IDH2 might be transcriptionally regulated by EZH2. In addition, we further inserted the promoter and 5’UTR of IDH2 (-1000 ~ 81 bp) into the pGL3-basic plasmid and performed a dual luciferase reporter assay after DZNep and GSK126 treatment in OVCAR8 and OVCAR4 cells. The transcriptional activity of the IDH2 promoter was attenuated upon treatment with DZNep but not GSK126 (Fig. 4E). Furthermore, we performed a dual luciferase reporter assay in OVCAR8-shEZH2#2 cells transduced with the EZH2- or EZH2-ΔSET-expressing plasmids. Doxycycline-induced EZH2 silencing decreased luciferase activity driven by the IDH2 promoter, whereas ectopic expression of EZH2 and EZH2-ΔSET restored IDH2 promoter activity (Fig. 4F). Moreover, we examined the binding regions of EZH2, SUZ12 and H3K27me3 in the IDH2 promoter by chromatin immunoprecipitation (ChIP). Compared with the IgG isotype control, the specific anti-EZH2 antibody but not the specific anti-SUZ12 or anti-H3K27me3 antibody bound directly to the region containing − 600~-400 bp in the IDH2 promoter (Fig. 4G). In addition, in our ChIP-Seq data and the GSE180448 data, the binding peaks of EZH2 but not those of H3K27me3 or SUZ12 were located in the promoter region of IDH2, providing further evidence supporting our hypothesis that EZH2 transcriptionally upregulates IDH2 via a noncatalytic function (Fig. 4H) [17, 26]. These results demonstrated that EZH2 serves as a direct transcriptional regulator to promote IDH2 expression in OC.
To confirm the regulatory effect of EZH2 on OXPHOS activity, we further examined the oxygen consumption rate (OCR) in OVCAR8 cells upon DZNep and GSK126 treatment. The basal and maximal OCRs were decreased upon treatment with DZNep but not GSK126 in OVCAR8 and OVCAR4 cells (Fig. 4I). Consistent with these findings, doxycycline induced EZH2 silencing also resulted in significant reductions in the basal and maximal OCRs in OVCAR8 cells (Fig. 4J). Furthermore, we demonstrated that the OCR of cells was restored after genetic knockdown of EZH2 and restoration with wild-type or mutant EZH2 (Supplementary Figure S2B). These data support a role for EZH2 in the regulation of OXPHOS in OC cells.
Reduction of EZH2 impairs IDH2 dependent metabolic rewiring to suppress OC growthTo investigate whether the downregulation of IDH2 mediated by EZH2 is clinically associated with OC, we performed IHC staining in our OC cohort and confirmed that IDH2 expression was positively correlated with EZH2 expression and that high expression of IDH2 predicted poor prognosis in OC patients (Fig. 5A-C). We further examined the colony formation and cell proliferation abilities upon silencing IDH2 or EZH2. Consistent with the above findings, silencing of either IDH2 or EZH2 was sufficient to suppress OC cell growth (Fig. 5D and E). We next examined the product synthesized via IDH2, α-ketoglutarate (α-KG), in OVCAR8 cells treated with DZNep or GSK126. The data indicated that the level of α-KG was significantly decreased upon treatment with DZNep but not GSK126 (Fig. 5F). We further evaluated the cellular α-KG level in cells after genetic knockdown of EZH2 and replacement with wild-type or mutant EZH2, demonstrating that the α-KG level was partially restored in OVCAR8 cells with Dox-induced EZH2 silencing by ectopic expression of EZH2 or EZH2-ΔSET (Fig. 5G). Moreover, when we supplemented cultures of OVCAR8 and OVCAR5 cells with α-KG after Dox-induced EZH2 silencing, the phenotype induced by EZH2 knockdown was partially rescued (Fig. 5H), suggesting that the oncogenic function of EZH2 in OC could be partially mediated by α-KG.
Fig. 5
Reduction of EZH2 impairs IDH2 dependent metabolic rewiring to suppress OC growth
(A) Representative images of EZH2 and IDH2 staining in OC tissues (n = 105). Scale bar: 100 μm
(B) IDH2 level was strongly positively correlated with EZH2 level in the OC cohort (n = 105). P value was determined by Pearson’s correlation test
(C) Kaplan-Meier survival analysis for the OC cohort (n = 105), which was divided into IDH2high and IDH2low subgroups based on the cutoff (cutoff = 5) calculated by the X-Tile software. The P value was calculated by log-rank test. **P < 0.01
(D) Colony formation assay in OVCAR8 and OVCAR4 cells transfected with IDH2 or EZH2 siRNA. Cells were cultured for 12 days. Growth medium was changed every 3 days. Images are representative of three independent experiments
(E) Cell growth curve of OVCAR8 and OVCAR4 cells transfected with IDH2 or EZH2 siRNA for 96 h. Data are presented as mean ± SD. *P < 0.05, **P < 0.01
(F-G) α-KG assay in OVCAR8 cells upon DNZep, GSK126 treatment (F) or in OVCAR8-shEZH2#2 cells ectopic expressing wildtype or mutant EZH2 (G). The OVCAR8 cells were treated with 2µM DZNep, 5µM GSK126 or 1 µg/mL doxycycline for 72 h before α-KG assay. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001
(H) Colony formation assay in OVCAR8 and OVCAR5 cells upon Dox-induced EZH2 silencing with or without α-KG supplementation. Cells were treated with 5ng/mL doxycycline for 12 days. Medium with or without 1mM α-KG was changed and doxycycline was also replenished every 2 days. Images are representative of three independent experiments
(I) Colony formation assay in OVCAR8-shEZH2#2 cells with ectopic IDH2 expression. Cells were treated with 5ng/mL doxycycline for 12 days. Growth medium was changed and doxycycline was also replenished every 3 days. Images are representative of three independent experiments. Immunoblot analysis of EZH2 and IDH2 levels in indicated samples. Cells were treated with or without Dox for 72 h before being harvested. GAPDH was used as loading controls
(J) Cell growth curve of OVCAR8-shEZH2#2 cells with ectopic IDH2 expression upon 1 µg/mL doxycycline treatment for 96 h. Data are presented as the mean ± SD. *P < 0.05
(K) Representative OCR pattern as a function of time (in min), normalized to total protein levels. Cells were incubated in the absence or presence of 1 µg/mL doxycycline for 72 h before OCR measurement. Data are shown as mean ± SD of three replicates per treatment
To examine whether the anticancer effect of EZH2 reduction is IDH2 dependent, we generated OVCAR8 cells stably expressing IDH2 and containing the EZH2 inducible knockdown construct shEZH2#2. IDH2 overexpression counterbalanced the downregulation of IDH2 mediated by doxycycline-induced EZH2 silencing (Fig. 5I). Next, we studied the tumorigenic behaviors of IDH2-overexpressing cells upon doxycycline induced EZH2 silencing. Ectopic expression of IDH2 partially rescued the reductions in colony formation and cell proliferation occurring upon doxycycline-induced EZH2 silencing (Fig. 5I and J). Furthermore, we found that the basal and maximum OCRs were restored in IDH2-overexpressing cells even upon doxycycline induced EZH2 silencing (Fig. 5K). Taken together, these results indicated that a reduction in EZH2 expression impairs IDH2 dependent metabolic rewiring to suppress OC growth.
Targeting EZH2-mediated metabolic rewiring exhibits potential therapeutic efficacy in preclinical models of OCTo further validate the role of EZH2-mediated metabolic rewiring in OC patients, we validated our findings in two primary OC cell lines derived from OC patients. Similarly, only the DZNep and YM281 inhibited the proliferation of patient-derived OC cells. Neither GSK126 nor EPZ-6438 exhibited growth inhibitory effects on in these patient-derived OC cell lines (Fig. 6A and B, Supplementary Figure S3A). We found that both DZNep and YM281 could reduce the levels of EZH2 and H3K27me3, whereas GSK126 and EPZ-6438 could only decrease only the level of H3K27me3 and did not affect the EZH2 protein levels in patient-derived OC cells (Fig. 6C).
Fig. 6
Targeting EZH2 mediated metabolic rewiring exhibits potential therapeutic efficacy in preclinical models of OC
(A) Colony formation assay in two primary OC cell lines derived from two OC patients (POVC17, POVC15). Cells were treated with DMSO, DZNep (1µM), YM281 (5µM), GSK126 (5µM) or EPZ-6438 (5µM) for 12 days. Growth medium was changed and inhibitors were also replenished every 3 days. Images are representative of three independent experiments unless stated otherwise
(B) Cell growth curve of two OC patient-derived primary tumor cell lines treated with DMSO, DZNep (1µM), YM281 (5µM) or GSK126 (5µM) for 96 h. Data are presented as the mean ± SD. ***P < 0.001
(C) Immunoblotting analysis of EZH2 and H3K27me3 levels in two OC patient-derived primary tumor cell lines upon EZH2 inhibitors treatment. Cells were treated with DMSO, DZNep (1µM), YM281 (5µM), GSK126 (5µM) or EPZ-6438 (5µM) for 72 h
(D) Tumor growth of PDX models in NOD-SCID mice treated with saline as control, EPZ-6438 at 200 mg/kg intragastrically daily, or DZNep at 1 mg/kg intraperitoneally twice a week (n = 6 per group). Data are presented as mean ± SD. ***P < 0.001(Two-way ANOVA with Tukey’s post hoc test)
(E) Bar graph showing the tumor weight on the 26th day post-treatment week (n = 6 per group). Data are presented as mean ± SD. **P < 0.01
(F) Body weight of each mouse post-treatment. Data are presented as mean ± SD.
(G) IHC analysis of EZH2, H3K27me3, IDH2 and Ki67 levels in the excised tumors from PDX models. Data are presented as mean ± SD. **P < 0.01, ***P < 0.001
(H) α-KG assay in PDX tumors upon EPZ6438 or DZNep treatment. Data are presented as mean ± SD. ***P < 0.001
Since chemoresistance has become a major obstacle in the treatment of OC, we established a patient-derived xenograft (PDX) model from a patient with chemoresistant (carboplatin- and Taxol-resistant) OC to study the efficacy of DZNep and EPZ6438 (Supplementary Figure S3B and C). The in vivo study indicated that DZNep but not EPZ-6438 significantly suppressed tumor growth (Fig. 6D and E). Treatment with either of these inhibitors resulted in a minimal body weight loss, suggesting that both inhibitors had low toxicity (Fig. 6F). In DZNep treated tumors, the Ki67 level was concomitantly decreased with that of EZH2 but not with that of H3K27me3, further indicating that the oncogenic role of EZH2 is independent of its catalytic activity (Fig. 6G and Supplementary Figure S3D). To further confirm that EZH2 inhibition can suppress the metabolic rewiring, we next examined the α-KG level in PDX tumors upon treatment with EZH2 inhibitors. The data showed that the α-KG level was significantly decreased upon DZNep treatment but not EPZ6438 treatment (Fig. 6H). The α-KG level is consistent with the levels of EZH2 and IDH2 (Fig. 6G and Supplementary Figure S3D), suggesting that α-KG is a key metabolite in OC that could predict poor prognosis. Collectively, these results demonstrated that inhibitors resulting in a reduction in EZH2 expression but not inhibition of its catalytic activity exhibit a potential therapeutic efficacy in patients with chemoresistant OC.
留言 (0)