
記住我
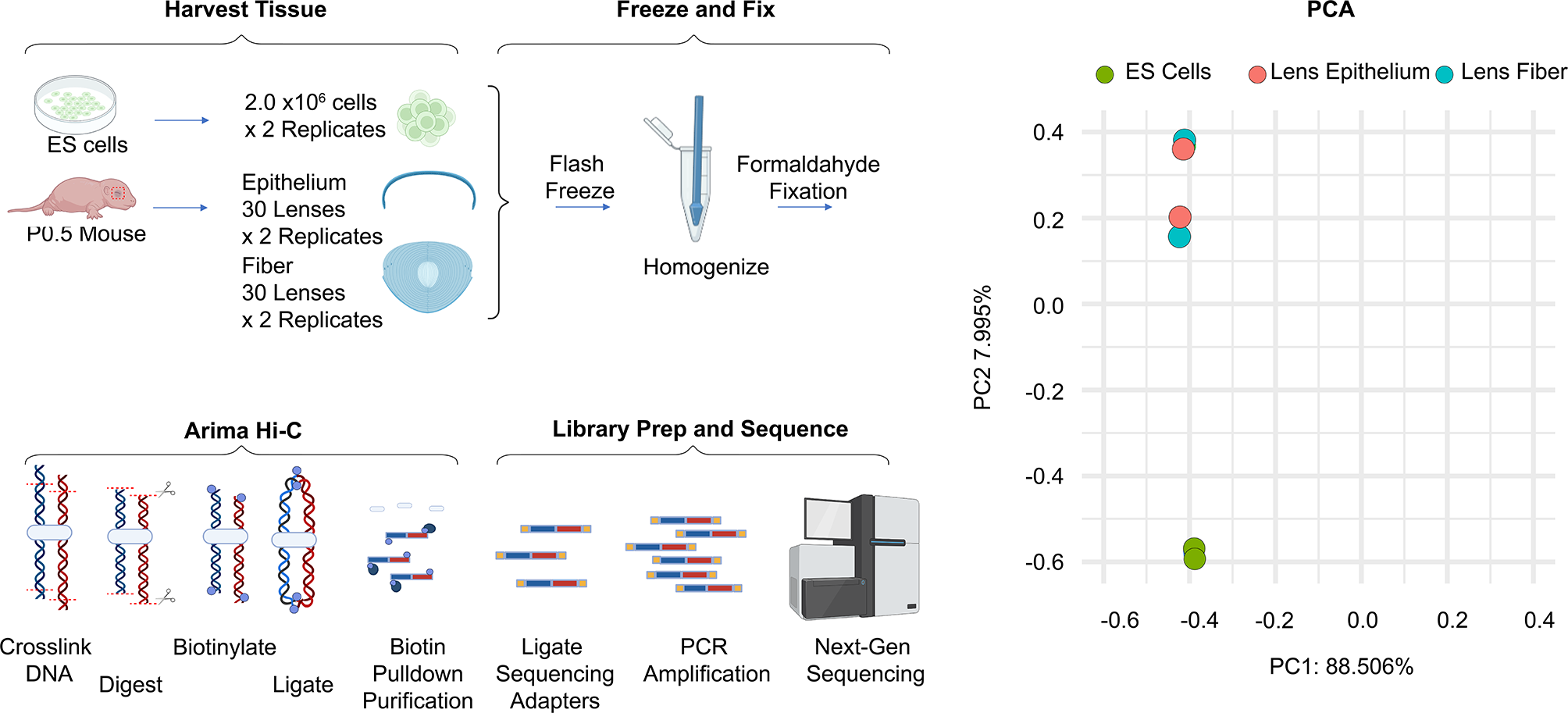


A schematic overview of sample multiplexing by NuHash is shown in Fig. 1. The detailed NuHash protocol is provided in Additional file 1. Isolated nuclei from cells (fresh or frozen samples) were stained with NuHash oligonucleotide-conjugated antibodies (NuHash antibody) and pooled before loading to the 10 × Genomics system to generate snATAC-seq libraries. Since the NuHash oligonucleotide was conjugated to the anti-Nuclear Pore Complex Proteins antibody and contains adapter sequences required for Illumina sequencing, the NuHash protocol does not require modifications to the 10 × Genomics library preparation or sequencing protocol. After sequencing, the number of NuHash reads per nucleus was counted using a Perl script (Additional file 3.pl). Based on the NuHash read counts, we demultiplexed the nuclei to the sample using a similar method developed for the Cell Hashing technique for scRNA-seq multiplexing [4].
Fig. 1
NuHash sample multiplexing schematic overview. Isolated nuclei from each sample are stained with a NuHash oligonucleotide-conjugated antibody containing a unique sample barcode. The stained nuclei from different samples were pooled for snATAC-seq library preparation, followed by massively parallel sequencing. Sequencing reads were then assigned to each nucleus using a nuclei (10x) barcode, and demultiplexing of nuclei from individual samples is carried out using the NuHash reads matching the sample barcode
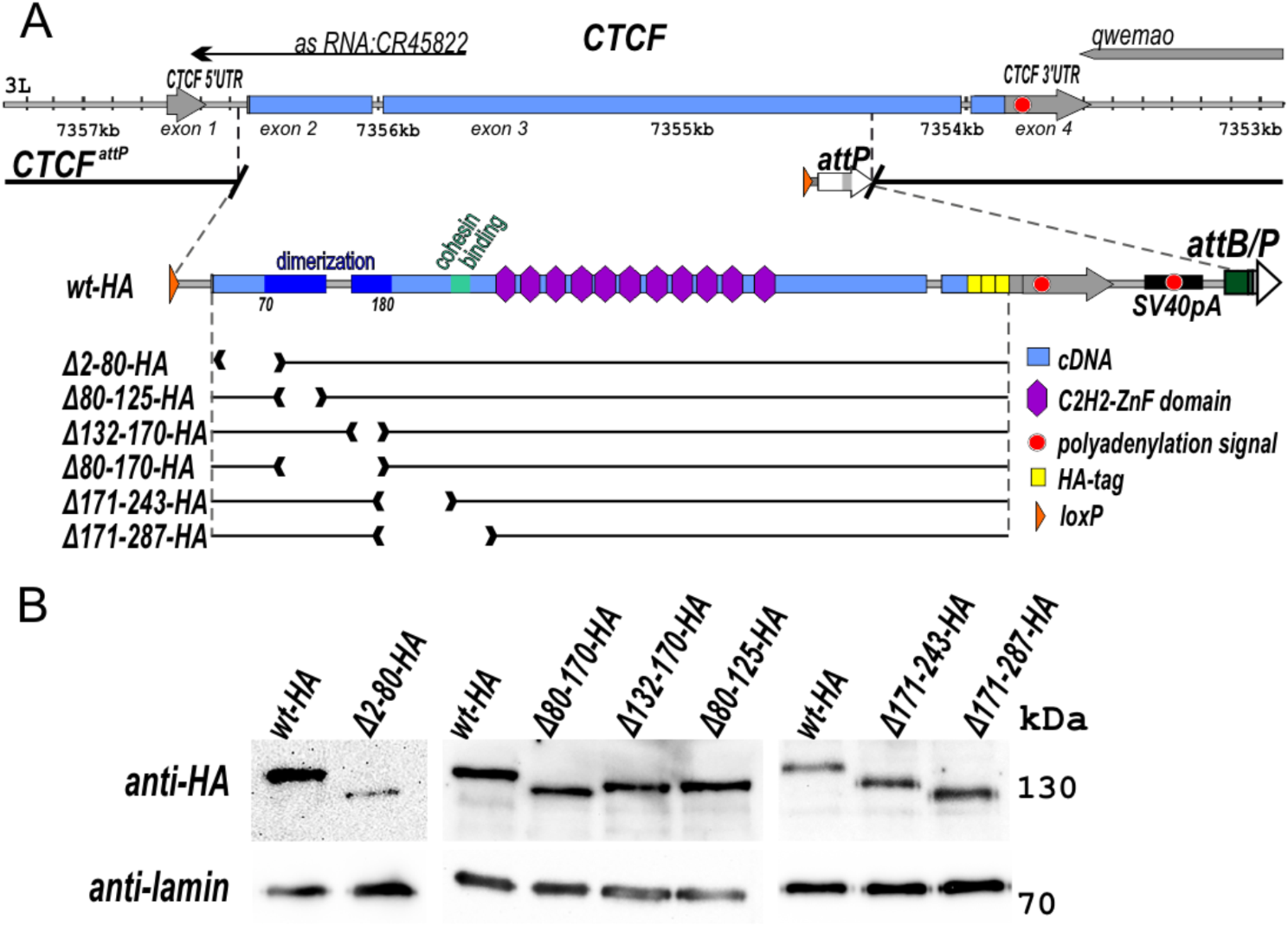
In Fig. 2, we illustrated a flow diagram with the molecular product progression through each step of our library preparation method. The left side shows the snATAC-seq products, and the right side shows the NuHash products. The NuHash oligonucleotide contains two unique molecular identifier sequences (UMI), the sample hashing sequence located between the UMIs, a part of the Illumina Read 1 sequence at the 5′ end, and a part of the Illumina Read 2 sequence at the 3′ end. We included two phosphorothioate bonds at the 3′ end of the sequence to protect the oligonucleotide probes from cell nucleases (Additional file 4: Table S1). The Illumina Read1 sequence binds to the gel beads during the Gel Beads-in-emulsion (GEM) step, and it acquires the complete sequence combination after amplification of the library.
Fig. 2
Key steps in NuHash library preparation. The snATAC-seq library preparation and NuHash library preparation are shown in the left and right panels, respectively. The NuHash oligonucleotide is composed of Illumina read1N (black), read2N (gray), two UMIs (yellow), and a sample-specific hash barcode (purple). The read1Ns are used for capturing by GEMs. The captured products are amplified linearly, during which the Illumina P5 adapter (blue) and 10 × barcode (green, a unique identifier for GEM) sequences are also added. Illumina P7 (orange) and sample index barcode (light gray) sequences are added to the products at the PCR amplification step. The full NuHash products contain full-length Illumina sequencing adaptors at both ends, 10 × barcode, two UMIs, NuHash barcode, and sample index barcode (bottom right)
Optimizing the ratio between hashing antibody and the number of nucleiThe NuHash oligonucleotide sequence contains Illumina P5 and P7 primer sequences, allowing us to amplify the NuHash products with the ATAC-seq products. This is one of the advantages of this method, and it helps to reduce the number of technical steps of library preparation. We tested different ratios of the NuHash antibody to the number of nuclei (Additional file 2: Fig. S1). We isolated nuclei from human CD4 + T cells and stained them with different concentrations of NuHash antibodies. As expected, the higher antibody ratio increased the amounts of NuHash products and reduced the amplification of the ATAC-seq products (Additional file 2: Fig. S1A). We observed that 0.01 µg of NuHash antibody per 50,000 nuclei consistently gave us clear ATAC-seq library characteristic banding patterns [1, 2] after removing large fragments (Additional file 2: Fig. S1B).
snATAC-seq library preparationWe performed two independent library preparations on mixtures of human and mouse nuclei to assess the accuracy of demultiplexing by alignment results. We used frozen human CD4 + T cells and a mouse hematopoietic progenitor cell line (HPC-7), and each sample was stained with a different NuHash antibody. For the first set, we used two samples: one human CD4 + T sample and one HPC-7 sample. For the second set, we used four samples in total, two samples of CD4 + T cells (male and female, allowing us to assess the accuracy of demultiplexing by alignment results) and two samples of HPC-7. Hereafter, we call the first set 2-plex and the second 4-plex. After staining with different NuHash antibodies, we evenly combined samples of nuclei and adjusted them to 7086 nuclei/sample/µl (2-plex) or 7340 nuclei/sample/µl (4-plex) to target 10,000–20,000 nuclei per sample. We assessed the quality of the libraries before sequencing by examining the fragment analyzer traces and observing the expected banding pattern of the fragment distributions [1, 2] (Additional file 2: Fig. s2).
Library sequencing and assessing qualities of the NuHash snATAC-seq librariesWe sequenced the libraries on the Illumina NextSeq 500 sequencer (50 bp for read1, 8 bp for i7 index read, 16 bp for i5 index read, and 50 bp for read 2). The paired-end sequence reads were aligned to a human (GRCh38) and mouse (mm10) combined reference genome (refdata-cellranger-atac-GRCh38-and-mm10-2020-A-2.0.0, 10xGenomics) using Cell Ranger ATAC software (version 2.0.0). The total numbers of read pairs and detected cells were 446,720,359 and 16,262 for 2-plex and 353,484,777 and 34,248 for 4-plex, respectively. The alignment statistics are summarized in Additional file 4: Table S2. We assessed the quality of the snATAC-seq libraries using ArchR [16]. From the ArchR analysis, we detected 2266 and 12,880 nuclei aligned to the human genome in 2-plex and 4-plex, respectively. The median fragment numbers per nucleus were 2103 (2-plex) and 1885 (4-plex), and the median transcription start site (TSS) enrichment was 22.69 (2-plex) and 22.808 (4-plex) (Additional file 2: Fig. S3A). The detected duplex was 51 of 2266 (2.3%, 2-plex) and 1658 of 12,880 (12.9%, 4-plex) (Additional file 2: Fig. S3B). In mouse alignment data, we detected 4360 and 13,054 nuclei aligned to the mouse genome in 2-plex and 4-plex, respectively. The median fragment numbers per nuclei were 2731 (2-plex) and 2280 (4-plex), and the median TSS enrichments were 21.09 (2-plex) and 22.69 (4-plex) (Additional file 2: Fig. S3C). The detected duplex was 0 of 4360 (0%, 2-plex) and 1,704 (13.1%, 4-plex) (Additional file 2: Fig. S3D). We observed the expected banding patterns in the insert fragment length distribution in all libraries (Additional file 2: Fig. S3E).
Accuracy of NuHashWe then selected nuclei with at least 10,000 read counts for further analysis, resulting in 10,574 nuclei (2-plex) and 12,208 nuclei (4-plex). We counted the number of hashing sequences per nucleus. We plotted the number of hash sequence counts for NuHash-1 (human) and NuHash-2 (mouse) using the 2-plex data (Fig. 3A). We observed a clear dissociation between NuHash-1- and NuHash-2-labeled nuclei based on the NuHash count status. In Fig. 3B, we plotted the number of reads aligned to mouse or human reference and colored them based on NuHash demultiplexing status. We assessed nuclear capture by testing the alignment rates of the reference sequence and the counts of NuHash per nucleus. We classified nuclei as singlet if the reads aligned to one of the reference genomes and doublet if the reads aligned to both. For NuHash, we classified nuclei as singlet if the NuHash reads aligned to a single NuHash barcode sequence and duplicate if the NuHash reads aligned to two or more different NuHash barcode sequences. We detected 9144 (86.48%) singlet nuclei and 1,251 (11.83%) duplicate nuclei, while 179 (1.69%) nuclei did not have sufficient NuHash counts (NA). Of the 9,144 singlet nuclei, 4409 (human/NuHash-1) and 4735 (mouse/NuHash-2) were classified based on the NuHash count status (Fig. 3A). We compared the nucleus-assigned classification (human singlet, mouse singlet, and duplicates) based on genome read alignments and NuHash count status. We detected only ten discordantly classified nuclei (8 human genome aligned nuclei with NuHash-2 and 2 mouse genome aligned nuclei with NuHash-1, p value < 2.2 × 10–16, chi-squared test). The calling accuracy in the 4-plex experiment was comparable to that in the 2-plex experiment (p value < 2.2 × 10–16, chi-squared test). We detected 12,208 singlets of those 7079 human nuclei with accurate NuHash information, 4979 mouse nuclei with accurate NuHash information, 974 newly classified nuclei, and only 150 nuclei with discordant classifications (Additional file 2: Fig. S4).
Fig. 3
Scatterplot showing the nuclear fragment alignment status of individual nuclei. A Based on read counts, nuclei are clustered into quadrants that are human-specific (bottom right, blue), mouse-specific (top left, purple), duplicated (top right, red), or low NuHash counts (bottom left, green). The black dotted lines indicate the cutoff for the counts used for NuHash demultiplexing. B Nuclei are colored by the NuHash demultiplexed status according to the ratios of the number of reads aligned to the human or mouse reference genomes
Clustering analysisWe performed single-cell clustering analysis to identify clusters of cells on human and mouse genome-aligned nuclei in the plex-4 experiment. We aligned the reads to a merged human and mouse reference genome (refdata-cellranger-atac-GRCh38-and-mm10-2020-A-2.0.0, 10xGenomics) to assess the alignment status of each nucleus. We eliminated the nuclei if one of the following conditions was met: (1) the percent of reads in peaks was less than 15% or (2) the number of fragments aligned to peaks was < 2000. A total of 1393 human nuclei and 2622 mouse nuclei passed these thresholds. Among those, we detected 5 clusters in mice (A, B, E, G, and H) and 4 in humans (C, D, F, and I) (Fig. 4A). As expected, a small number of hashed mouse nuclei were clustered with the human nuclei cluster (1.5%) and vice versa (6.3%) (Fig. 4B). To further assess the classification accuracy, we aligned the reads to human (refdata-cellranger-arc-GRCh38-2020-A-2.0.0, 10 × Genomics) or mouse (refdata-cellranger-arc-mm10-2020-A-2.0.0, 10 × Genomics) reads independently. We eliminated the nuclei if one of the following conditions was met: 1) the percent of reads in peaks was less than 15%, 2) the ratio of reads aligned to the genomic blacklist (loci with anomalous, unstructured, or high signal in next-generation sequencing experiments independent of the cell line or experiment [17]) was > 0.05, 3) nucleosome signal was < 0.2 or > 4, and 4) the number of fragments aligned to peaks was < 2000. We analyzed differentially open chromatin regions (OCRs) between Human 1 (female) and Human 2 (male). We identified 29 differential OCRs between male and female human samples (Additional file4: Table S3). The top 4 male-specific OCRs (9 OCRs) were located on chromosome Y. We also found that 5 female-specific OCRs (20 OCRs) were on chromosome X (Additional file4: Table S3). We plotted the top female-specific OCR, which is in the gene body of transmembrane protein 191B (TMEM191B) (Fig. 4C), and the top male-specific OCR, which is located in the transcription start site of zinc finger Y-chromosomal protein (ZFY) (Fig. 4D), as a representation of sample hashing accuracy. We detected a peak at TMEM191B in only Human 1 (female) and at ZFY in only Human 2 (male). This result supports the high demultiplexing accuracy of NuHash.
Fig. 4
snATAC-seq analysis identified distinct nucleus clusters by open chromatin status. A The nuclei were visualized by UMAP and colored by cluster (five mouse and four human clusters). The letters represent the individual samples (or subjects). B UMAPs similar to A but plotted separately for each sample. The numbers indicate the detected nuclei in mouse (top) or human clusters (bottom). C and D Sample-specific read alignment and peak detection at selected genes. C In the gene body of TMEM191B, only reads of the human 1 (female) sample were detected. D In contrast, only reads of the human 2 (male) sample were detected at the promoter region of the human ZFY gene in chromosome Y
Bulk ATAC-seqTo assess the accuracy of our NuHash approach, we performed ultradeep bulk ATAC-seq on HPC7 mouse cells to obtain a well-defined open-chromatin region (OCR) profile of the cell line. A total of 8 ATAC-seq libraries from independent cell culture batches were sequenced with the goal of obtaining > 50 million paired-end reads per sample. We excluded one sample that had fewer than 10,000 paired reads. The sequencing statistics of each library are summarized in Additional file 4: Table S4. We obtained a median of 69.8 million paired-end reads per sample (a total of 700.7 million paired-end reads used in the analysis), with a median percentage of reads in OCRs of 58.8%, a median percent of duplication of 0.22%, and detected OCRs ranging from 84,593 to 132,852 (mean = 105,983, standard deviation = 17,024).
Peak characteristics by the number of cell subtype clusters in which the peak was presentWe assessed the characteristics of snATAC-seq peaks categorized by the number of cell subtype clusters (clusters) in which the peak was present. A higher number of clusters indicates that the peaks were constitutively present, and a lower number means the peaks were cell subtype specific. To increase the robustness of the analysis, we reperformed clustering only for mouse nuclei and selected clusters containing at least 50 nuclei (Fig. 5A). We identified 85,951 peaks and four clusters (A, B, C, and D) in the mouse HPC7 snATAC-seq dataset. Among the identified peaks, 77,987 overlapped with OCRs detected in bulk ATAC-seq (see previous section). Of these 77,987 peaks, 16,810 peaks were detected in only one cluster (Cnum_1), 11,964 in two clusters (Cnum_2), 12,743 in three clusters (Cnum_3), and 36,470 in all four clusters (Cnum_4). When we then looked at the peak intensity of the overlapped bulk ATAC-seq OCRs, they were positively correlated with the number of clusters in which the peak was present (Fig. 5B). The constitutive peaks (Cnum_4) were enriched in promoters and 5’UTR regions (Additional file 2: Fig. S5). Interestingly, the peak heights of the Cnum_4 bulk ATAC-seq OCRs were bimodally distributed, suggesting the existence of stochastic OCRs (low-intensity peaks) and constant OCRs (high-intensity peaks) (Fig. 5B). The high-intensity peaks were enriched in promoters and 5’UTR regions, while the low-intensity peaks were enriched in gene bodies. This finding suggests that the molecular mechanisms that create constitutively open chromatin differ between stochastic and constant OCRs (Fig. 5C).
Fig. 5
Integration analysis of snATAC-seq and deeply sequenced bulk ATAC-seq highlights a relationship between peak intensity and peak genomic locations. A UMAP shows the mouse nuclei identified in the snATAC-seq analysis. B Peak intensities of overlapping bulk ATAC-seq peaks were plotted according to the numbers of cell clusters in which the peaks were found in the snATAC-seq analysis. C The distribution of peaks over different genomic features detected in all clusters dichotomized by the peak intensities of overlapping bulk ATAC-seq peaks. High-intensity peaks were most prevalent in exonic regions and promoters, while lower-intensity peaks were most common in the gene body and intergenic regions
We also assessed the gene expression and transcriptional regulatory properties of each peak category group. While the GENCODE genes (Mouse Release M15) containing the Cnum_4 high-intensity peak group had higher expression levels than the low-intensity peak group or other Cnum groups containing genes (Additional file 2: Fig. S6A), transcriptionally active putative enhancers with potential bidirectional transcription (TAPEs) [18] (Additional file4: Table S5) evenly overlapped with both the low- and high-intensity peak groups (Additional file 2: Fig. S6B). A comparison of motif enrichment analysis results between low- and high-intensity groups revealed that the CTCF/BORIS motif was significantly enriched in the low-intensity peak group (Additional file 2: Fig. S6C). The proportions of overlap with CTCF ChIP-seq peaks (CTCF peaks) were significantly higher in the Cnum_4 groups (low-intensity, 41.9%, chi-square statistic = 4038.4864, p < 0.00001; high-intensity, 40.2%, chi-square statistic = 2105.6447. p < 0.00001) than all peaks detected in bulk ATAC-seq (22.14%) (Additional file 2: Fig. S6D). The absolute distances of the CTCF peaks from the TSSs were significantly shorter in the high-intensity peak group than in the low-intensity peak group or all bulk ATAC-seq peaks (Additional file 2: Fig. S6E). Since CTCF and cohesins are master regulators of topologically associating domains (TADs), we also tested whether these CTCF peaks were located in promoter-interacting regions (PIRs) [19, 20]. While only 12.7% of CTCF peaks in low-intensity groups overlapped with PIRs, 60.4% of CTCF peaks in high-intensity groups overlapped with PIRs, suggesting that functional CTCF peaks might be enriched in high-intensity groups.
留言 (0)