
記住我
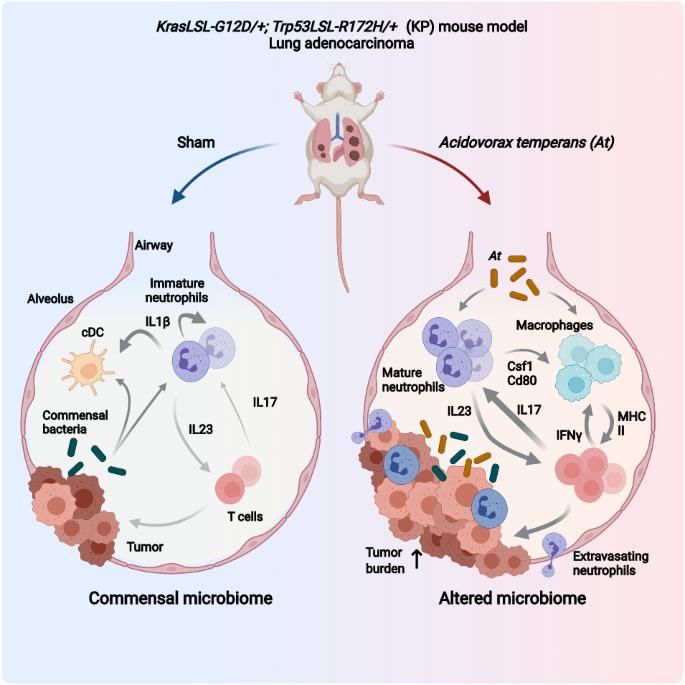


Considering our previous findings that PELI1 showed higher expression in breast cancer than adjacent tissues, we examined whether PELI1 is commonly abundant in other cancers by a multiple tumor tissue microarray assay, including colon cancer, lung cancer, rectum cancer, breast cancer and prostate cancer. Interestingly, comparable high-expression of PELI1 was observed in these cancers relative to breast cancer, suggesting that PELI1 is an epigenetically regulated pan-cancer oncogene (Fig. S1A, B). To explore the underlying mechanisms involved in the breast cancer progression mediated by PELI1, we performed co-immunoprecipitation assays for PELI1 in MDA-MB-231 cells followed by mass spectrometry (Fig. 1A). Many candidates of binding partners of PELI1 were identified, which were significantly enriched into distinct signaling besides posttranslational modification (Fig. 1B and Table S1). Notably, EGFR was also identified by its six specific peptides, and involved in most of these enriched signaling (Fig. S1C), indicating that EGFR is one of the interacting proteins of PELI1.
Fig. 1: The levels of PELI1 and EGFR were positively correlated in human breast cancer.
A MDA-MB-231 cells overexpressing PELI1 were lysed and immunopurified with normal IgG and anti-PELI1 antibody respectively. Then the complex was resolved by SDS–PAGE followed by Coomassie Blue staining. The distinct bands were analyzed by MS. Red arrows indicated the identified PELI1 and EGFR. B Representative biological processes and signaling pathways significantly enriched from proteins identified from Co-IP with PELI1 in MDA-MB-231 cells. GO and KEGG analysis were performed using DAVID bioinformatics database. The number of enriched proteins in relative terms was shown in each bar. C, D IHC analysis of PELI1 and EGFR in the tissues from breast cancer patients’ tissues microarray. Representative images of IHC staining (C) and the correlation rate (D) by Pearson’s test is shown (N = 121, scale bar, 50 μm). E Overall survival rates were determined by Kaplan–Meier analyses of indicated groups. Hazard ratio (HR) and P values (log rank P) are shown. F Western blotting analysis of PELI1 protein levels in MDA-MB-231 cells with EGFR knockdown. GAPDH was used as loading control. G ELISA analysis of the change of PELI1 in MDA-MB-231 cells with Gefitinib. H Western blotting analysis of EGFR protein levels in MDA-MB-231 cells with PELI1 knockdown. GAPDH was used as a loading control. I Sample immunoblotting showed the levels of PELI1 and EGFR proteins in mammary gland from indicated transgene mice. β-Tubulin was used as a loading control.
Given that EGFR is closely related to the tumorigenesis, we analyzed the PELI1 and EGFR expression using the GEPIA database, and found that PELI1 was positively correlated with EGFR expression in multiple cancers besides breast cancers (Fig. S1D). We also examined the PELI1 and EGFR protein levels in breast cancer tissue microarray by IHC, and found a highly positive correlation (Fig. 1C, D). Furthermore, the highest co-expression was also detected in breast cancer cells among five kinds of cancer cell lines, including MDA-MB-231, HCT-116, H1299, SKVO3 and HT-29 cells (Fig. S1E). Notably, breast cancer patients with both high expressions showed a lower survival rate than the ones harboring their single high or both low expressions (Fig. 1E).
To further confirm their correlations, we silenced EGFR expression with siRNA, and found that PELI1 protein levels were dramatically reduced after EGFR knockdown in MDA-MB-231 cells (Fig. 1F). Furthermore, the expression of PELI1 was also markedly suppressed by EGFR inhibitors such as Gefitinib and Lapatinib, as indicated by the immunoblotting and enzyme-linked immunosorbent assay (Figs. 1G and S1F, G). These data indicated that EGFR regulates the expression of PELI1.
We next examined whether PELI1 reversely regulates EGFR expression. PELI1 knockdown with shPELI1 lentivirus led to a remarkable decrease of EGFR in MDA-MB-231 cells (Fig. 1H). Furthermore, EGFR expression was further evaluated in PELI1 knockout background. PELI1flox/flox mice were mated with MMTV-Cre mice to generate mammary conditional knockout mice (CreMMTV; PELI1f/f). As expected, EGFR expression were markedly decreased in mammary tissue from CreMMTVPELI1f/f mice compared to WT group (Fig. 1I). These results confirmed that PELI1 is positively correlated with EGFR expression.
PELI1 physically interacted with EGFRConsistent with the findings from IP-MS, we also found that PELI1 was closely colocalized with EGFR by immunofluorescence staining in MDA-MB-231 cells (Fig. 2A, B). To further confirm the interaction between PELI1 and EGFR, we transfected pcDNA3.1-HA-PELI1 and pLVX-FLAG-EGFR constructs into HEK293T/17 cells, followed by the immunoprecipitation with anti-HA and anti-FLAG antibodies respectively. The Co-IP results showed that PELI1 and EGFR were reciprocally coimmunoprecipitated with each other, indicating a physical interaction between both of them (Fig. 2C). This is further supported by the Co-IP assays of endogenous PELI1 and EGFR in MDA-MBA-231 cells (Fig. 2D).
Fig. 2: PELI1 directly interacted with EGFR.
A, B IF staining showed co-localization between PELI1and EGFR in MDA-MB-231 cells (scale bar, 10 μm). The co-localization correlation rate of PELI1 and EGFR in A is shown (B). C The interaction between PELI1 and EGFR was detected by Co-IP assay. HEK293T/17 cells were co-transfected with pCDNA3.1-HA-PELI1 and pLVX-FLAG-EGFR plasmids. The cells were harvested and subjected to immunoprecipitated with anti-HA and anti-FLAG antibodies respectively. Similar Co-IP analysis with normal IgG were performed as control. D Co-IP analysis of endogenous PELI1 and EGFR in MDA-MB-231 cells. The cell extracts were immunoprecipitated with anti-IgG, anti-EGFR and anti-PELI1 antibodies, respectively, upon EGF (100 ng/ml) stimulation. E The sketch map of the deletion mutant regions of PELI1. F Co-IP analysis of PELI1 mutants binding to EGFR. HEK293T/17 cells were co-transfected with PELI1 deletion mutants (HA tagged) and pLVX-FLAG-EGFR plasmids, and IP analysis was performed with anti-FLAG antibody. G The sketch map of the deletion mutant regions of EGFR. H Co-IP analysis of EGFR regions binding to PELI1. HEK293T/17 cells were co-transfected with EGFR deletion mutants (FLAG-tagged) and pCDNA3.1-HA-PELI1 plasmids, and IP analysis was performed with ani-HA antibody. I GST-pull down assay of the direct correlation between PELI1 and EGFR-TK.
To determine the domains that are responsible for their interactions, we constructed a series of truncated forms of EGFR and PELI1 (Fig. 2E, G). Correlatively, EGFR showed a specific interaction with FHA domain of PELI1 (Fig. 2F), while the intracellular tyrosine kinase (TK) domain of EGFR is responsible for the interaction with PELI1 (Fig. 2H). This was further confirmed by the pull-down assays (Fig. S2), which showed that TK-domain-truncated EGFR (His tag) were specifically retained in the presence of PELI1 (Fig. 2I).
PELI1 stabilized EGFR through K63-linked ubiquitinationConsidering PELI1 as an E3 ubiquitin ligase and its interaction with EGFR, we investigated whether EGFR is a potential target for PELI1. We first performed cycloheximide (CHX) treatment in MDA-MB-231 cells transfected with shScramble and shPELI1 lentivirus, respectively. CHX, an inhibitor of protein synthesis, led to a mild decrease of EGFR in MDA-MB-231 cells. On the contrary, PELI1 knockdown accelerated the time-dependent decrease of EGFR proteins, indicating that PELI1 protects EGFR from the degradation (Fig. 3A, B).
Fig. 3: PELI1 ubiquitinated EGFR and inhibited its degradation.
A, B Western blotting analysis of EGFR protein levels in MDA-MB-231 cells with PELI1 knockdown upon the treatment of CHX (10 μg/ml). EGFR levels were normalized to the change of GAPDH (N = 3). C Western blotting analysis of K63-linked polyubiquitination of EGFR immunoprecipitated from HEK293T/17 cells co-overexpressing PELI1 and EGFR upon EGF stimulation (100 ng/ml). D Western blotting analysis of EGFR proteins in MDA-MB-231 cells with or without PELI1 overexpression upon EGF (100 ng/ml) stimulation. E Flow cytometric analysis of membrane EGFR levels in MDA-MB-231 cells with PELI1 overexpression upon EGF (100 ng/ml) stimulation (N = 3). ***P < 0.001. F Effect of PELI1 knockdown on the cell viability of MDA-MB-231 cells was detected upon the treatment of Gefitinib.
We next defined the type of polyubiquitinated linkages attached to EGFR mediated by PELI1. K48-linked ubiquitination are the most common chain type and target proteins for proteasomal degradation, while K63-linked ubiquitination has many well-studied non-degradative roles on the substrates [33, 34]. HEK293T/17 cells were transfected with HA-tagged K63-linked ubiquitin and FLAG-tagged EGFR combined with PELI1 overexpression, followed by the immunoprecipitation for EGFR and subsequent immunoblotting analysis of its ubiquitination. Interestingly, overexpression of PELI1 promoted K63-linked ubiquitination of EGFR but it failed to detect any increase of K48-linked ubiquitination of EGFR upon enforced expression of PELI1 (Figs. 3C and S3A, B). Furthermore, we found that enforced expression of PELI1 led to the increase of EGFR in MDA-MB-231 cells (Fig. 3D). The plasma membrane EGFR was also upregulated as indicated by the flow cytometry analysis (Fig. 3E).
EGFR plays roles in malignant transformation and cancer metastasis, and its dysregulated activation has been regarded as multifaceted hallmarks of cancer cells [35, 36]. Thus, we further examined the contributions of PELI1 to aberrant EGFR signaling in cancers. As expected, Gefitinib, an inhibitor of EGFR approved for the clinical treatment of cancers [37], induced a dose-dependent reduction of the cell viability in MDA-MB-231 cells, and this is attenuated by the enforced expression of PELI1 (Fig. S3C). On the contrary, PELI1 knockdown enhanced the sensibility of EGFR inhibitor against MDA-MB-231 cells (Fig. 3F). To further confirm the gains of PELI1 inhibition in the EGFR-targeting therapy, we performed similar cell viability assays in two lung cancer cell lines NCI-H1650 and NCI-H1975 with oncogenic activations of EGFR. In line with the findings in breast cancers, EGFR expression is correlated with the highly expressed PELI1 in these two lung cancer cell lines (Fig. S3D). Similarly, PELI1 knockdown also enhanced the sensibility of EGFR inhibitor against NCI-H1650 and NCI-H1975 (Fig. S3E, F). In addition, immunoblotting demonstrated that EGFR signaling and EMT related proteins (Vimentin and SNAIL) were considerably downregulated after PELI1 knockdown in these two lung cancer cell lines (Fig. S3G). Taken together, these data demonstrated that PELI1 mediated EGFR stability through K63-linked ubiquitination to be involved in the aberrant EGFR signaling in cancers.
EGFR activation led to the phosphorylation and activation of PELI1To further determine the regulatory effect of EGFR on PELI1, we evaluated the expression of PELI1 upon EGF stimulation in MBA-MB-231 cells. The immunoblotting showed that PELI1 protein levels were significantly increased with EGFR activation indicated by the upregulated phosphorylation (Fig. 4A), which coinciding with an increase of the plasma membrane EGFR (Fig. S4A). Notably, the rapid upregulation of PELI1 expression in MBA-MB-231 cells after 5 min EGF stimulation, suggested that EGFR regulated the stability of PELI1 proteins.
Fig. 4: EGFR phosphorylated PELI1 leading to its K63-linked auto-ubiquitination.
A Western blotting analysis of the levels of indicated proteins in response to EGF (100 ng/ml). B, C Western blotting analysis of the tyrosine (B) and threonine (C) phosphorylation of PELI1 with EGFR overexpression upon EGF stimulation (100 ng/ml) in MDA-MB-231 cells. D, E Western blotting analysis of the tyrosine or threonine phosphorylation of PELI1 immunoprecipitated from HEK293T/17 cells with overexpression of the indicated PELI1 mutants. F Western blotting analysis of the K63-mediated polyubiquitination of PELI1 immunoprecipitated from HEK293/17 cells co-overexpressing with EGFR and PELI1 upon EGF (100 ng/ml) stimulation. G Similar with F excluding overexpressing PELI1 mutant (K169F). H PELI1 was immunoprecipitated from HEK293T/17 cells transfected with PELI1, PELI1-Y154F or PELI1-T264A plasmids, followed by Western blotting analysis of the K63-mediated polyubiquitination. I Western blotting analysis of EGFR and phospho-EGFR in MDA-MB-231 cells overexpressed PELI1 or PELI1-T264A upon EGF (100 ng/ml) stimulation. J Flow cytometric analysis of membrane EGFR in MDA-MB-231 cells with overexpression of PELI1, PELI1-Y154F or PELI1-T264A (N = 3). **P < 0.01, ***P < 0.001. All P values were determined by unpaired two-tailed Student’s t test.
Given that the phosphorylation of PELI1 is required for its activation and subsequent autoubiquitylation [38], we thus analyzed the phosphorylation status of PELI1 upon EGF stimulation in MBA-MB-231 cells. Endogenous PELI1 proteins were immunoprecipitated from MBA-MB-231 cells, followed by immunoblotting for the phosphorylated tyrosine residues and threonine residues respectively (Fig. 4B, C). The elevated levels of phosphorylated Tyr/Thr in MBA-MB-231 cells with EGFR overexpression and activation indicated that EGFR is an upstream phosphokinase of PELI1. We then performed phosphoamino acid analysis and identified three novel phosphorylation sites of PELI1, including Tyr154, Thr175 and Thr264 (Fig. S4B, C).
To further define the phosphorylation sites of PELI1, we constructed three corresponding inactive mutants of PELI1, containing a single mutation as Thr175 (T175A) and Thr264 to alanine (T264A), and Tyr154 to phenylalanine (Y154F) respectively. Interestingly, Y154F mutant failed to show EGFR-induced increase of phosphorylated Tyr/Thr compared to that of WT form of PELI1 (Fig. 4D). Furthermore, T264A mutant but not T175A completely abolished the phosphorylation of Thr upon the EGFR activation (Fig. 4E). These data suggested that PELI1 was phosphorylated at Tyr154 upon EGFR activation that led to its phosphorylation at Thr264.
We then determined whether PELI1 undergoes the autoubiquitination after the phosphorylation induced by EGFR. Indeed, overexpression of EGFR promoted K63-linked ubiquitination of PELI1 (Fig. 4F). However, it failed to show any K48-linked ubiquitination of PELI1 upon enforced expression of EGFR (Fig. S4D). Additionally, we also identified the ubiquitination site of PELI1 as Lys 169 via IP-MS analysis (Fig. S4E, F). To further confirm it, we transfected a mutant form PELI1 (K169R) and EGFR into HEK293T/17 cells and immunoblotting demonstrated that this lysine residue is mainly responsible for the K63-linked ubiquitination of PELI1 (Fig. 4G).
In line with the previous study to show the roles of the phosphorylation of PELI1 in its activation [38], we found that overexpression of PELI1-Y154F, as well as PELI1-T264A, reversed the K63-linked ubiquitination of PELI1 mediated by EGFR (Fig. 4H). Indeed, overexpression of PELI1-T264A also failed to show the increase of intracellular EGFR and the plasma membrane EGFR in MDA-MB-231 cells (Fig. 4I, J). Therefore, these results revealed that EGFR activation led to the phosphorylation of PELI1 at Y154 residue that is required for the activation of PELI1.
Inhibition of PELI1 and EGFR suppressed breast cancers metastasisEMT is required for cancer metastasis, and has closely relationship with EGFR inhibitor resistance [39]. Thus, we next characterized the functional roles of the relationship between PELI1 and EGFR in the EMT progress. In keeping with previous studies, either PELI1 knockdown or Gefitinib treatment inhibited the ability of the migration, invasiveness and tumor spheres formation of MDA-MB-231 cells, which were further strongly enhanced by the combined treatments (Figs. 5A–C and S5A-C). Consistently, knockdown of PELI1 led to additional reduce of EMT-related proteins compared to the treatment of Gefitinib, including Vimentin, SNAIL and SLUG (Fig. 5D). The co-inhibition of PELI1/EGFR also led to a dramatic decrease of Vimentin and increase of E-cadherin, as indicated by immunofluorescence staining (Fig. S5D).
Fig. 5: Inhibition of PELI1 and EGFR suppressed breast cancer.
A–C Quantification of the migration (A) and invasion (B, C) of MDA-MB-231 cells transfected with PELI1-shRNA with or without Gefitinib (2 μM) treatment (N = 3, scale bar, 100 μm). D Western blotting analysis of the indicated proteins with or without PELI1 knockdown and Gefitinib (2 μM) treatment. E–G Effects of PELI1 knockdown and Gefitinib treatment on the tumor incidence of MDA-MB-231 cells in nude mice. The mice were subcutaneously transplanted with MDA-MB-231/Con-shRNA and MDA-MB-231/PELI1-shRNA cells (5 × 106/mouse) and were treated with or without Gefitinib (50 mg/kg) orally every other day for 2 months. Representative images of tumors (E) and tumor weight (F) are represented (N = 6 per group). The tumors were made into paraffin sections and the Ki67-positive cells (G, scale bar, 50 μm) were quantified. H Effects of PELI1 knockdown and Gefitinib treatment on the lung-metastasis of MDA-MB-231 cells. NYG mice were injected with MDA-MB-231/Con-shRNA and MDA-MB-231/PELI1-shRNA cells (2 × 105/mouse) via tail vein, and were treated with or without Gefitinib (50 mg/kg) orally every other day for 1 month. The whole indicated lung tissues were stained with Bouin fluid and made into HE stained sections (scale bar, 50 μm). I Quantitative analysis of the metastatic lung nodules in H (N = 6 per group). J The GFP fluorescence intensity of lung tissues from H are shown. *P < 0.05, **P < 0.01, ***P < 0.001. All P values were determined by unpaired two-tailed Student’s t test.
Next, we used the mouse subcutaneous xenograft model to evaluate the effect of combination of PELI1 knockdown and EGFR inhibition on tumor growth. As expected, single inhibition of PELI1 and EGFR led to smaller tumors than controls. Notably, their combination led to further marked reduction in tumors growth (Fig. 5E, F). Furthermore, the immunohistochemical staining of the tumors demonstrated that Ki67 expression, a well-known proliferative marker, was dramatically down-regulated in the tumors from the mice with the combined treatments of PELI1 knockdown and Gefitinib (Fig. 5G).
The efficacy of this combination against breast cancers was further confirmed by major reductions in metastatic capacity of cancer cells in vivo. we found that combination of PELI1 knockdown and EGFR inhibition significantly decreased metastatic potential of cancer cells to lung in caudal vein xenograft models compared to the either single treatment, as demonstrated by reduced number of micrometastatic nodules (Fig. 5H). Similarly, considerable reduce of MDA-MB-231 cells-derived tumor nodules in lung were also detected in mice with both PELI1 knockdown and EGFR inhibition (Fig. 5I), as further confirmed by the decreased GFP fluorescence intensity indicating the MDA-MB-231 cells number in lung (Fig. 5J). Together, these results demonstrated that PELI1 knockdown synergized with EGFR inhibitor and that combined therapy showed superior activity against breast cancer metastasis.
The compound S62 interrupted the interaction between PELI1 and EGFR to suppress breast cancer metastasisBased on the decrease of PELI1 protein indicated by ELISA assay in MDA-MB-231 cells (Fig. S6A), we screened our in-house small-molecule library of 200 compounds and identified S62 as a potential small molecule disruptor of PELI1/EGFR (Fig. 6A). Compared to the co-immunoprecipitation with each other in control group cells, S62 treatment impaired the interaction of PELI1 and EGFR (Fig. 6B). Indeed, both protein levels of PELI1 and EGFR were greatly reduced after S62 treatment in MDA-MB-231 cells (Figs. 6C and S6B). S62 treatment also led to a decrease of the membrane EGFR (Fig. 6D), whereas it failed to block the phosphorylation of EGFR upon EGF stimulation (Fig. 6E). Furthermore, S62 reduced the tyrosine and threonine phosphorylation of PELI1 and K63-linked ubiquitination of EGFR (Fig. 6F, G).
Fig. 6: The compound S62 interrupted the interaction between PELI1 and EGFR to suppress breast cancer metastasis.
A Chemical structure of S62. B HEK293T/17 cells with overexpression of PELI1 and EGFR were treated with or without S62 (5 or 10 μM) for 24 h. Co-IP assay was subsequently performed to detect the interaction between PELI1 and EGFR. C Western blotting analysis of EGFR and PELI1 in MBA-MB-231 cells with the treatment of S62 for 24 h. D Flow cytometric analysis of the membrane EGFR in MDA-MB-231 cells treated with or without S62 for 24 h (N = 3). E Western blotting analysis of EGFR, PELI1 and phosphorylation of EGFR with or without the treatment of S62 (10 μM) upon EGF stimulation (100 ng/ml). F Western blotting analysis of the tyrosine and threonine phosphorylation of PELI1 immunoprecipitated from MDA-MB-231 cells. The cells were treated as in E. G Western blotting analysis of the K63-mediated polyubiquitination of EGFR immunoprecipitated from HEK293T/17 cells with the treatment of S62 (10 μM). H Quantitative analysis of the migration of MDA-MB-231 cells with the treatment of S62 (N = 3). I Effect of S62 on the invasion of MDA-MB-231 cells. The representative images of invasive cells were shown (scale bar, 100 μm). J Quantitative analysis of the invasion of MDA-MB-231 cells in I (N = 3). K Western blotting analysis of E-cadherin and SNAIL in MDA-MB-231 cells with the treatment of S62. L The representative images of lung metastasis of breast cancer cells in NYG mice with S62 treatment. MDA-MB-231 transfected with lentivirus that stably expressed luciferase (2 × 105/mouse) were injected into NYG mice via the tail vein. The mice were treated with CMC-Na (0.5%) or S62 (10 or 50 mg/kg) every day for 2 weeks, and then were detected using the bioluminescence imaging. *P < 0.05, ***P < 0.001. All P values were determined by unpaired two-tailed Student’s t test.
Next, we evaluated the effects of S62 against breast cancers. Similar with the findings from the combination of PELI1 knockdown and EGFR inhibition, S62 significantly inhibited the migration and invasiveness of MDA-MB-231 cells (Figs. 6H–J and S6C). Moreover, immunoblotting demonstrated that S62 treatment led to a dose-dependent increase of E-cadherin protein level but a decrease of SNAIL (Fig. 6K). We then examined the MDA-MB-231 cells-derived tumor nodules in lung after caudal vein injection, and found that S62 inhibited the metastasis of MDA-MB-231 cells as indicated by the decreased luciferase intensity in lung (Fig. 6L).
Indeed, S62 showed mild effect on the cell viability of tumor cells or normal cells (Fig. S6D–K) and even the MDA-MB-231 cells sensitivity to Gefitinib (Fig. S6L). On the other hand, overexpression of PELI1 did not reverse the suppressed migration of MDA-MB-231 cells caused by S62 treatment (Fig. S6M). These results indicated that S62 functions as a disruptor of PELI1 and EGFR interaction but not binding to PELI1 or EGFR alone.
留言 (0)