
記住我
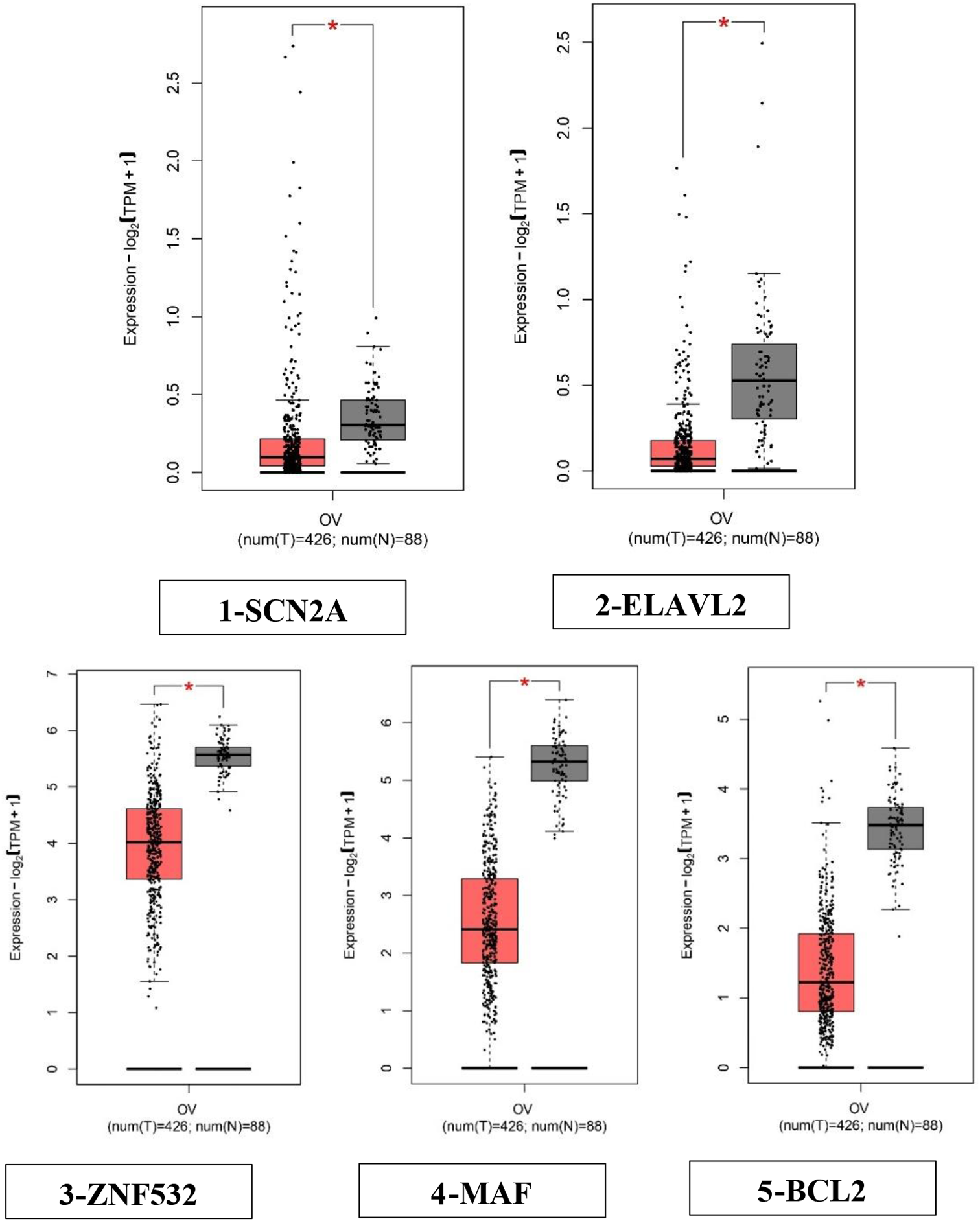

A total of 51 pyroptosis-associated genes (PYAGs) were finally identified and included in this study. Their potential biological functions regarding pyroptosis and molecular regulation mechanisms are summarized in Fig. 1A. We show somatic mutations and copy number variation (CNV) of these 51 PYAGs in 436 ovarian cancers from the TCGA-OV database with a waterfall diagram. A relatively high mutation frequency observed in this TCGA-OV cohort (Fig. 1B). A total of 400 (91.97%) among 436 samples experienced mutations in PYAGs (Fig. 1B). TP53 (88%) exhibited the highest mutation frequency, followed by NLRP3 (3%), NLRP2 (3%) NOD1 (2%), NLRP6 (1%), GSDMB (1%), NLRC4 (1%), NOD2 (1%), TP63 (1%), CASP5 (1%), IRF1/2 (all 1%), NLRP1/7 (all 1%), PLCG1(1%), CASP1/6/8 (all 1%), CHMP4C (1%), PPKACA (1%), and TNF (1%), while the other PYAGs did not show any mutations in these OC samples (Fig. 1B). As TP53 showed the highest mutation frequency, we explored the relationship between TP53 mutation and PYAGs expression. The results showed that expression levels of 2 (NLRC4 and IL1A) among 51 PYAGs were significantly associated with TP53 mutation status (Additional file 2: Fig. S2A-B). As to CNV in PYAGs, GSDMD, GSDMC, TP63, CHMP6, PRKACA, NLRP3, TNF, AIM2, PJVK, BAK1, GZMB, CASP4, CASP5, CASP1, PYCARD, CASP8, CHMP4C, NLRC4, CHMP4B, and CHMP2B showed widespread CNV amplifications, while CASP9, NLRP7, NLRP2, CHMP2A, CASP3, IRF2, CASP6, NLRP6, GZMA, BAX, PLCG1, ELANE, GPX4, and CHMP7 showed prevalent CNV deletions (Fig. 1C). The locations of CNV alterations of 51 PYAGs on their respective chromosomes were shown in Fig. 1D. Furthermore, principal component analysis (PCA) based on expression of these 51 PYAGs in TCGA-OV and GTEx samples revealed that PYAGs could completely distinguish OV samples from normal samples (Fig. 1E-F). We further analyzed DNA methylation levels of 51 PYAGs with DiseaseMeth 2.0, the results showed AIM2, CASP1, CASP8, GSDMC and NLRP6 exhibited significantly lower methylation levels compared with those in normal control group (Additional file 2: Fig. S2C-G and Additional file 9: Table S4).
We further investigated mRNA expression levels of PYAGs between OC and normal samples in TCGA-OV and GTEx database, and found a positive correlation between mRNA expression and CNV alterations in most of PYAGs. Compared to normal ovarian tissues, the mRNA expression levels of PYAGs with CNV gain was markedly increased in OC tissues, such as GSDMC, CHMP6, NLRP3, TNF and AIM2, while PYAGs with CNV loss showed lower expression in tumors, such as CASP9, CHMP7, ELANE, PLCG1 and BAX. However, some PYAGs with CNV gain, such as GSDMD, TP63, PRKACA, PJVK and CASP4, showed downregulated mRNA expression in ovarian cancers, and some PYAGs with CNV loss, such as IL18, GPX4, GZMA, NLRP6 and CASP6, showed upregulated mRNA expression in ovarian cancers (Fig. 1F). Thus, while CNV can explain many observed changes in PYAGs expression, CNV is not the only factor involved in the regulation of mRNA expression. The above findings indicate the high heterogeneity of genomic and transcriptomic alteration landscape in PYAGs of OC patients, suggesting that pyroptosis might play a crucial role in OC development and progression.
Fig. 1
Landscape of genetic alterations and transcriptional patterns of PYAGs in ovarian cancer. A Summary of the potential biological functions of pyroptosis and their molecular regulation mechanism. B Genetic alterations of 51 PYAGs in 436 patients with ovarian cancer from TCGA-OV cohort. C CNV variation frequency of 51 PYAGs in ovarian cancer. Red dot represents the amplification frequency; blue dot represents the deletion frequency. D The location of CNV alteration of 51 PYAGs on 23 chromosomes. E Principal component analysis (PCA) for the expression of 51 PYAGs to distinguish ovarian cancer from normal samples in TCGA-OV cohort and GTEx data. Red dot: ovarian cancer samples; Blue dot: normal ovarian samples. F Expression levels of 51 PYAGs between ovarian cancer and normal ovarian tissues from TCGA-OV and GTEx cohorts. *P < 0.05; **P < 0.01; ***P < 0.001. PYAG: pyroptosis-associated genes; OV: ovarian cancer; TMB: tumor mutation burden; CNV: copy number variation; PCA: Principal component analysis
Identification of pyroptosis-associated subtypes in OCFirstly, we explored prognostic value of PYAGs in patients across 33 cancer types. We found that all PYAGs were associated with overall survival of patients in at least one cancer type (Additional file 2: Fig. S2H). Subsequently, a total of 1332 ovarian cancer samples from six eligible OC cohorts (TCGA-OV, GSE140082, GSE63885, GSE32062, GSE26193, and GSE17260) were enrolled for further analysis. Kaplan–Meier analysis and univariate Cox regression revealed the prognostic values of 49 PYAGs in these OC patients, and P < 0.05 was selected as the threshold for filtering (Additional file 2: Fig. S2I and Additional file 9: Table S5). The comprehensive landscape of PYAGs interactions, molecular connections, and their prognostic significance in patients with OC was demonstrated in a pyroptosis network (Fig. 2A and Additional file 9: Table S6). Furthermore, through Spearman’s correlation analysis, we found a strong relationship of the 51 PYAGs with the TME-infiltrating immune cells utilizing the GSEA algorithm in TCGA-OV database. Most of these PYAGs were positively correlated with the immune cells in varying degrees. However, six PYAGs (CASP9, CHMP7, NLRP1, PJVK, PLCG1 and PRKACA) was negatively related to almost all these immune infiltration cells (Fig. 2F). The above results indicated that cross-talk among different PYAGs may play critical roles in the formation of different pyroptosis patterns and TME cell-infiltrating characterization between individual tumors.
To further explore the involvement of pyroptosis in OC, a consensus clustering algorithm was employed to stratify the patients based on the qualitatively different expression of these 49 genes (Additional file 3: Fig. S3A-H). Accordingly, k = 2 was identified as an optimal selection for clarifying the entire samples into two pyroptosis-associated clusters (PAC) including PAC1 (n = 515 cases) and PAC2 (n = 819 cases) (Fig. 2B). Principal component analysis (PCA) analysis revealed significant differences in the pyroptosis transcription profiles between the two distinct clusters (Fig. 2C), and patients in PAC1 showed significant survival advantage compared with those with PAC2 (P = 0.001, Fig. 2D). Furthermore, as shown in Fig. 2E, Patients in PAC2 were preferentially related to advanced stage (FIGO III-IV) and poor differentiation (histological grade 3) compared to patients in PAC1 (P < 0.05).
Fig. 2
Consensus clustering to identify PACs and their correlation with clinicopathological and biological characteristics. A Interaction among PYAGs in ovarian cancer shown by network. B Two PACs (k = 2) and their correlation areas calculated by consensus clustering algorithm. C Differences in the transcription profiles between the two PACs analyzed by PCA. D Survival difference between the two PACs shown by Kaplan-Meier curves based on five GEO cohorts (GSE140082, GSE17260, GSE26193, GSE32062, and GSE63885) and TCGA-OV cohort. E Differences in clinicopathologic features, expression levels of PYAGs between two PACs in OC cohorts with heatmap. F Correlation between 51 PYAGs and immune cells in the gathered TCGA-OV cohort by heatmap. G Activated biological pathways in two PACs visualized by heatmap through GSVA enrichment analysis. *P < 0.05; **P < 0.01; ***P < 0.001. PYAG: pyroptosis-associated genes; PAC: pyroptosis-associated cluster; OV: ovarian cancer; PCA: Principal component analysis; GSVA: Gene Set Variation Analysis; OS: overall survival
TME infiltration characteristics in distinct PACsTo explore the biological behaviors related to the two distinct PACs, we conducted GSVA enrichment analysis against the Hallmarker gene set. As shown in Fig. 2G and Additional file 9: Table S7, PAC1 was markedly enriched in immune fully-activated pathways, including inflammatory response, interferon gamma response, natural killer cell mediated cytotoxicity, T cell and B cell receptor signaling pathway as well as NOD-like and Toll-like receptor signaling pathways, cell adhesion and JAK-STAT signaling pathways (Fig. 2G). While PAC2 was prominently associated with immune suppression biological process (Fig. 2G). Accordingly, subsequent analysis of TME immune cell infiltration with ssGSEA indicated that PAC1 showed higher level of infiltration of most immune cells than PAC2, including activated CD4+/CD8+T cells, activated B cells, natural killer cell, macrophage, eosinophil, mast cell and plasmacytoid dendritic cell (Fig. 3A). What’s more, we utilized ESTIMATE algorithm to evaluate immune infiltration (stromal score, immune score, and ESTIMATE score) and tumor cell purity (Tumor Purity) (Additional file 9: Table S8) of the tumors with PAC1 versus those with PAC2, which further demonstrated that PAC1 displayed higher immune scores, stromal score and ESTIMATE score compared with PAC2, and PAC2 showed a higher tumor purity than PAC1 (Fig. 3B-D), suggesting that abundant nontumor components (e.g., immune cells and stromal cells) were existed in ovarian cancer with PAC1. We also characterized the infiltration of 22 human immune cells in OC between the two clusters with CIBERSORT, which demonstrated obviously higher infiltration of activated memory CD4+ T cells, activated NK cells, M1 macrophages, T gamma delta cells, activated mast cells and neutrophils in the PAC1 compared to PAC2, while M0 and M2 macrophages, naive B cells, and regulatory T cells (Tregs) were significantly enriched in the PAC2 (Fig. 3E). Furthermore, the expression of immune checkpoints, such as PD-L1, PD-1, CTLA4 and LAG3, were significantly upregulated in PAC1 than those in PAC2 (Fig. 3F-I). These findings suggest that the two clusters had significantly distinct TME cell infiltration characterization.
Fig. 3
Characterization of TME infiltration characteristics in the two PACs. A Abundance of 23 tumor-infiltrating immune cells in two PACs using the ESTIMATE algorithm (Kruskal-Wallis H test). B,C TME score and tumor purity of different PACs analyzed with vioplot. D Correlation between TME score and tumor-infiltrating immune cells of two PACs by pheatmap. E Fraction of tumor-infiltrating lymphocyte cells in two PACs with CIBERSORT algorithm. F-I Expression levels of PD-L1, PD-1, CTLA4 and LAG3 between two PACs. *P < 0.05; **P < 0.01; ***P < 0.001. PYAG: pyroptosis-associated gene; TME: tumor microenvironment; ESTIMATE: Estimation of Stromal and Immune Cells in Malignant Tumors using Expression Data
Gene subtypes based on pyroptosis phenotype-related DEGs in OCAlthough the PYAGs were classified into two clusters by consensus clustering algorithm in OC patients, the underlying genetic alterations and potential biological behavior within these clusters remains to be clarified. A total of 4342 overlapping DEGs were identified between two PACs. GO enrichment analysis showed that these DEGs were significantly enriched in biological processes related to immune regulation, such as T cell activation and leukocyte cell − cell adhesion (Additional file 3: Fig. S3I-K and Additional file 9: Table S9). KEGG analysis demonstrated DEGs were enriched in cytokine − cytokine receptor and tumor-related pathways (Additional file 3: Fig. S3L-N and Additional file 9: Table S9), suggesting that pyroptosis exerts a nonnegligible function in the immune regulation of the TME. We then employed Univariate Cox regression analysis to screen out 889 DEGs with significant favorable overall survival (OS) of OC patients (all P < 0.05) (Additional file 9: Table S10). Based on the transcriptional levels of 889 pyroptosis-related gene signatures, unsupervised consensus clustering algorithm was performed to obtain three distinct pyroptosis gene subtypes which were identified as gene cluster A (534 cases), gene luster B (465 cases), and gene cluster C (335 cases), respectively (Fig. 4A and Additional file 4: Fig. S4A-H). PCA analysis confirmed discernible dimensions among the transcription profiles of the three gene clusters (Fig. 4B). Survival analysis showed that patients in gene cluster C (335 patients) showed the worst survival outcome among the three subtypes (P < 0.001) (Fig. 4C). Furthermore, gene cluster C was associated with advanced FIGO stage and grade 3, and patients in PAC1 and gene cluster A showed favorable overall survival (Fig. 4D), which indicated that three distinct gene clusters were characterized by different clinicopathologic feature and survival outcome. Moreover, three gene clusters displayed significant differences in the expression of 49 PYAGs (Fig. 4E).
To explore the potential role of the pyroptosis-related gene signatures in the TME immune infiltration, we analyzed the expression of immune checkpoints, chemokine, cytokines and other factors among three gene clusters of OC, and found that the expressions of immune checkpoints (PD-L1, CTLA-4, and LAG3), chemokines (CXCL10, CCL5, and CXCL13), interleukins interferons (IFNG, IFNB1, and IFNAR2) and MHC molecules (HLA-A, HLA-B, and HLA-C) [26] were significantly upregulated in gene cluster A and B compared with those in gene cluster C (Fig. 4F and Additional file 4: Fig. S4I-L), suggesting that gene cluster A and B was regarded as immune activated characteristic. However, gene cluster C showed higher expression of molecules (TGF-β2 and Smad9) related with TGF-β/EMT pathway than gene cluster A and B (Additional file 4: Fig. S4M), indicating gene cluster C was deemed as stromal activated characteristic and tumor promotion.
Fig. 4
Construction of gene clusters based on PACs-related DEGs. A Identification of three gene clusters (k = 3) based on PACs-related DEGs by consensus clustering algorithm, gene cluster A (534 patients), gene cluster B (465 patients), and gene cluster C (335 patients). B The remarkable difference among transcriptome profiles of three gene clusters with PCA. C The OS of three gene clusters in TCGA and five GEO cohorts shown by Kaplan-Meier curves (log-rank tests, P < 0.001). D Relationships between clinicopathologic features and three gene clusters analyzed by unsupervised clustering. E Differences in the expression of 49 PYAGs among three gene clusters. F Differences in the expression levels of immune checkpoints among three gene clusters in TCGA and five GEO cohorts. *P < 0.05; **P < 0.01; ***P < 0.001. DEGs: Differentially expressed genes; PCA: Principal component analysis; OS: Overall survival
Construction of Pyrsig score and exploration of its clinical relevanceGiven that the crucial regulation function of PYAGs in prognostic evaluation and TME immune landscapes, we further constructed a scoring system with PCA algorithm, termed as Pyrsig score, to quantify pyroptosis regulation and immune microenvironment in individual OC sample. We showed that low Pyrsig score (59% alive and 41% dead) presented prominent survival advantage than high Pyrsig score group (42% alive and 58% dead) (P < 0.001, Fig. 5A, B), indicating that Pyrsig score showed potentially prognostic value of OC patients. More importantly, significant differences in Pyrsig score were observed among different PACs (Fig. 5C) and three gene clusters (Fig. 5D), gene cluster A showed the lowest Pyrsig score while gene cluster C presented the highest Pyrsig score (Additional file 9: Table S11) (all P < 0.001). The distribution of samples in two PACs (PAC1 and PAC2), three gene clusters (gene cluster A/B/C), and two Pyrsig score groups (low and high score) were illustrated with alluvial diagram (Fig. 5E), indicating that gene cluster C with PAC2 displayed a higher Pyrsig score, showing worse survival outcome, whereas gene cluster A with PAC1 exhibited a lower Pyrsig score with favorable prognosis (Fig. 5E). Additionally, Pyrsig score showed no statistic difference between serous ovarian cancer and others types in OC (P > 0.05, Additional file 5: Fig. S5A-B) and displayed statistical significance among different grades (G1-2 vs. G3) (P < 0.001, Additional file 5: Fig. S5C-D) and FIGO stages (I-II vs. III-IV) (P < 0.01, Additional file 5: Fig. S5E-F). Survival status in patients with serous ovarian cancer (Additional file 5: Fig. S5G), G1-2(Additional file 1: Fig. S5I), G3 (Additional file 5: Fig. S5J), stage III-IV (Additional file 5: Fig. S5L) all displayed statistical significance.
Increasing evidence has demonstrated that somatic mutation patterns were associated with responsiveness to immunotherapy [38]. We found no difference in tumor mutation burden (TMB) between the low and high Pyrsig score groups (Additional file 5: Fig. S5M). However, Kaplan-Meier analysis showed that patients in high TMB group showed better survival outcome than those in low TMB group (P = 0.024, Fig. 5F). Through combination of Pyrsig scores and TMB, we revealed that in high Pyrsig score group, the survival rate of patients with high TMB was higher than that of patients with low TMB (log-rank test, P = 0.005, Fig. 5G). We then analyzed the distribution variations of somatic mutations between the two Pyrsig score groups in TCGA-OV cohort. The top ten mutated genes were TP53, TTN, MUC16, CSMD3, TOP2A, NF1, USH2A, HMCN1, RYR2, and FAT3 (Fig. 5H, I). Patients with a high Pyrsig score showed higher frequencies of USH2A, TOP2A and FLG mutations compared to those with a low Pyrsig score. However, opposite effect was observed regarding the mutation levels of TP53, TTN, and MUC16 (Fig. 5H, I). Moreover, we assessed potential correlation between Pyrsig score and Cancer Stem Cell (CSC) index in OC, and found that CSC index (RNAss and DNAss) showed no significant correlation with Pyrsig score in OC (both P > 0.05) (Additional file 5: Fig. S5N-O), indicating that OC cells with different Pyrsig score possessed no distinct stem cell properties and cell differentiation.
Fig. 5
Construction of Pyrsig score and exploring the relationship between Pyrsig score and clinical features. A, B The relationship between survival outcome and Pyrsig score in patients from TCGA and five GEO cohorts (Log-rank test, P < 0.001). C, D Level of Pyrsig score in two PACs and three gene clusters (Kruskal-Wallis H test, P < 0.01). E Distributions of two PACs, three gene clusters, Pyrsig scores and survival outcomes in OC from TCGA-OV and five GEO cohorts with Alluvial diagram F Survival analysis of patients with low and high TMB in TCGA-OV cohort with Kaplan-Meier. G Difference in prognostic advantages among four groups stratified by Pyrsig score and TMB in TCGA-OV cohort shown by Kaplan-Meier curves (Log-rank test, P = 0.005). H, I Somatic mutation features established with low and high Pyrsig scores by waterfall plot. H-TMB: High tumor mutation burden; L-TMB: Low tumor mutation burden
留言 (0)