Patients
A cohort of 422 patients with breast cancer and 69 pairs of patients with primary breast foci with metastatic lesions were recruited at the Shengjing Hospital of China Medical University from April 2011 to July 2013. Patients with breast cancer were diagnosed based on radiological and pathological examinations. Individual breast cancer patients were excluded if they had incomplete clinical data, received neoadjuvant chemotherapy or radiotherapy, another type of malignant tumor, severe organ dysfunction, or bilateral breast cancer. Surgical tumor tissues were collected, fixed in 10% formalin, and paraffin-embedded for histological examination and immunohistochemistry (IHC). Written informed consent was obtained from all patients. The experimental protocol was approved by the Institutional Research Ethics Committee of Shengjing Hospital of China Medical University (Project ID 2018PS304K, approved on 03/05/2018).
Histology
The paraffin-embedded tissue Sects. (4 μm) were dewaxed and rehydrated, followed by hematoxylin and eosin (H&E) staining. The sections were photographed under a light microscope (Olympus, Tokyo, Japan) and examined independently by two pathologists in a blinded manner.
Immunohistochemistry
Tissue Sects. (4 μm) were dewaxed, rehydrated, and treated with 3% H2O2 in methanol, followed by antigen retrieval in citrate buffer (pH 6.0) in a microwave for 10 min. After being blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline with Tween® 20 (TBST), the sections were probed with mouse anti-SOST (ab63097, Abcam, Cambridge, MA, USA) at 25 ℃ for 2 h. The bound antibodies were detected using horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobulin IgG at room temperature for 30 min and visualized using 3,3′-diaminobenzidine, followed by counterstaining with hematoxylin. The sections were mounted and photographed under a light microscope. The intensities and frequencies of positively stained cells were evaluated using IHC Profiler software [19].
Cell culture
Human breast cancer MDA-MB-231, MCF-7 cells, and mouse osteogenic precursor cells (MC3T3-E1) were obtained from the American Type Culture Collection (Manassas, VA, USA). SCP2 cells were a gift from Professor Joan Massague (Cell Biology Program and Howard Hughes Medical Institute, Memorial Sloan-Kettering Cancer Center, NY, USA). MDA-MB-231 cells were cultured in Leibovitz’s L15 medium (Thermo Fisher, Carlsbad, CA, USA), and MCF-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% of fetal bovine serum (FBS, Biological Industries, Cromwell, CT, USA). MC3T3-E1 cells were cultured in α-MEM plus 10% of FBS and 1% P/S. All cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.
Transduction
SCP2 cells (107/well) were transduced with lentivirus at a multiplicity of infection of 10 for the expression of control shRNA or SOST-specific shRNA (with green fluorescent protein (GFP), Sangon Biotech, Shanghai, China) in the presence of 5 µg/mL puromycin (A1113803, Thermo Fisher) for 7 days to establish stable SCP2/NC and SOST-knockdown SCP2/KD cells. The efficacy of SOST silencing was determined by Western blotting. The shRNA target sequences were: SOST KD1, 5’-GCAGTGAAAGATGTAGCCAAA-3’ and SOST KD2, 5’-GCCTCAGATAATCTGGTGAAA-3’. We retrieved the SOST gene sequence from GenBank and designed the primers: SOST-F (EcoRI): AGGGAGACCCAAGCTGGCTAGTTGaattcGCCACCATGCAGCTCCCACT, SOST-R (BamHI): GTCACTTAAGCTTGGTACCGAggatccGTAGGCGTTCTCCAGCTCGGC. In addition, the cDNA fragment of SOST was cloned into pcDNA3.1-CMV-MCS-3flag-EF1-ZsGreen-T2A-Puro vector and sequenced. MDA-MB-231 cells were transfected with control plasmid or the SOST-expressing plasmid and selected by treatment of cells with G418 to establish stable SOST over-expressing MDA-MB-231 (MDA-MB-231/OE) cells or control MDA-MB-231/NC cells. The levels of SOST expression were quantified by Western blot.
Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from SCP2/KD, SPC2/NC, MDA-MB-231/OE or MDA-MB-231/NC cells using TRIzol reagent (Thermo Fisher). The RNA samples were reverse transcribed into cDNA. The relative levels of gene mRNA transcripts to the control GAPDH were quantified by qRT-PCR using PrimeScript RT Master Mix (RR047A, Takara, Kyoto, Japan) and TB Green Premix Ex Taq II (RR820A, Takara), according to the manufacturer’s instructions. Data were analyzed by 2−ΔΔCt.
Western blotting
MDA-MB-231, SCP2, MCF-10A, SCP2/NC, and SCP2/KD cells were lysed in radioimmunoprecipitation assay (RIPA) buffer and centrifuged (1000 × g at 4 °C for 20 min). After determining protein concentrations using a bicinchoninic acid assay kit (Thermo Fisher), the cell lysates (40 µg/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels and transferred to polyvinylidene fluoride membranes (MilliporeSigma, Burlington, MA, USA). The membranes were blocked with 5% BSA in TBST and incubated with primary antibodies at 4 °C overnight. The primary antibodies included anti-SOST (ABIN6997488, antibodies-online GmbH, Germany), anti-TGF-β (ER31210, HuanBio, Hangzhou, China), anti-SMAD3 (66,516–1-1 g, PTG, Rosemont, USA), anti-CXCR4 (60,042–1-1 g, PTG), anti-STAT3 (10,253–2-AP, PTG), anti-p-STAT3 (9138, Cell Signaling Technology), anti-KRAS (12,063–1-AP, PTG), and anti-β-actin (20,536–1-AP, PTG). The bound antibodies were detected using HRP-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). The immune signals were visualized using enhanced chemiluminescence reagent (Thermo Fisher). The relative levels of individual target proteins to β-actin were determined by densitometric analysis using ImageJ software (US National Institutes of Health, Bethesda, MD, USA).
Immunoprecipitation
SCP2 cells were harvested and lysed with cold RIPA lysis buffer containing protease inhibitors, followed by centrifuging. The cell lysates (50 µg/tube) were reacted with anti-SOST, anti-STAT3, or control isotype IgG (2 µg) with gentle agitation at 4 ºC overnight. In addition, the cells lysates (1 ml) were pre-treated with 200 μM S6 compound at room temperature for one hour and reacted with each type of antibodies. The generated immunocomplexes were precipitated with 20 µl of protein A/G plus-agarose beads (sc-2003, Santa Cruz, USA) at 4 ºC for 4 h. After being centrifuged, the palleted beads were washed with TBST and the bound proteins were eluted with 2 × SDS loading buffer. The eluted proteins were analyzed by SDS-PAGE and stained with anti-STAT3 or anti-SOST to detect the direct interaction of SOST with STAT3.
Enzyme-linked immunosorbent assay (ELISA)
SCP2 cells were co-cultured with MC3T3-E1 cells (5 × 105 cells/well) in 6-well plates and treated with DMSO or S6 compound for 24 h. The levels of CXCL12 in the supernatants of cultured cells were determined using an ELISA kit (KE10049, PTG) and 3,3',5,5'-tetramethylbenzidine, according to the manufacturer’s instructions. The experimental samples were tested in triplicate. The absorbance of each well was measured at 450 nm using a microplate reader (Biotek, USA). The CXCL12 concentrations were calculated using a standard curve established with recombinant CXCL12 protein provided.
Chemotaxis assay
Breast cancer cells (3 × 104/chamber) were suspended in 2% of FBS medium (200 µl) and cultured in the top chamber of 24-well transwell plates (3422, Corning, USA). The bottom chamber was cultured with MC3T3-E1 cells up to 80% confluence in 200 µl of medium containing 2% of FBS. The top and bottom chambers were co-cultured for 24 h. The cells on the upper membrane of the top chamber were removed using a cotton ball while the cells that migrated the bottom surface of the top chamber membrane were stained with 0.5% (v/m) crystal violet and counted in a blinded manner.
Adhesion assay
To mimic the bone matrix, MC3T3-E1 cells were induced for osteogenic differentiation in MEM-α media containing 10% of FBS, 10 mM β-glycerophosphate (Solarbio), and 50 µg/ml of ascorbic acid (Solarbio) for 9 days. Subsequently, the cultured MC3T3-E1 cells were treated with 20 mM NH4OH (Sigma, USA) and 0.5% Triton X-100 (Solarbio) for 5 min to form the bone matrix layer. GFP expressing breast cancer cells (2 × 105 cells/well) were added into each bone matrix layer and incubated for 15 min. After aspirating floating cells, the adhering cells were washed with PBS and counted under a fluorescent microscope.
Cell viability and cytotoxicity assay
Viability and cytotoxicity were analyzed using a Cell Counting Kit-8 (CCK8) (Dojindo, Japan), according to the manufacturer’s instructions. SCP2/WT, SCP2/NC, SCP2/KD1, SCP2/KD2, MDA-MB-231 and MCF-7 cells were cultured in 96-well plates for 24, 48 or 72 h in the presence or absence of different concentrations of S6 compound or positive controls of 50 μM epirubicin (EADM) or 5 nM docetaxel (DTX). During the last 4-h culture, the cells were exposed to 10% CCK8 solution. The absorbance at 450 nm in individual wells was measured using a microplate reader (Biotek, USA).
Biolayer interferometry (BLI)
The affinity of S6 binding to SOST was measured, as described previously [20] using an Octet K2 instrument (ForteBio). All assays were run at 30 ºC with continuous shaking of 1000 RPM using the assay buffer of 0.1% BSA, 0.01% Tween-20 and 1% DMSO in PBS. SOST (10,593-H07H, SinoBio, Beijing, China) at 0.15 mg/ml was dissolved in sterile water, biotinylated and immobilized onto the Octet SSA biosensors. S6 (C29H24N6O2S, molecular weight 520.62, Chemdiv, USA) was dissolved in PBS and adjusted to different concentrations for BLI. After each round of association and disassociation, the SOST-contained sensors were washed with the assay buffer for 10 min to remove nonspecifically bound molecules. Raw kinetic data were obtained. The kon and koff values were analyzed using the software provided and the Kd values were calculated, based on double reference subtraction.
Murine xenograft tumor model
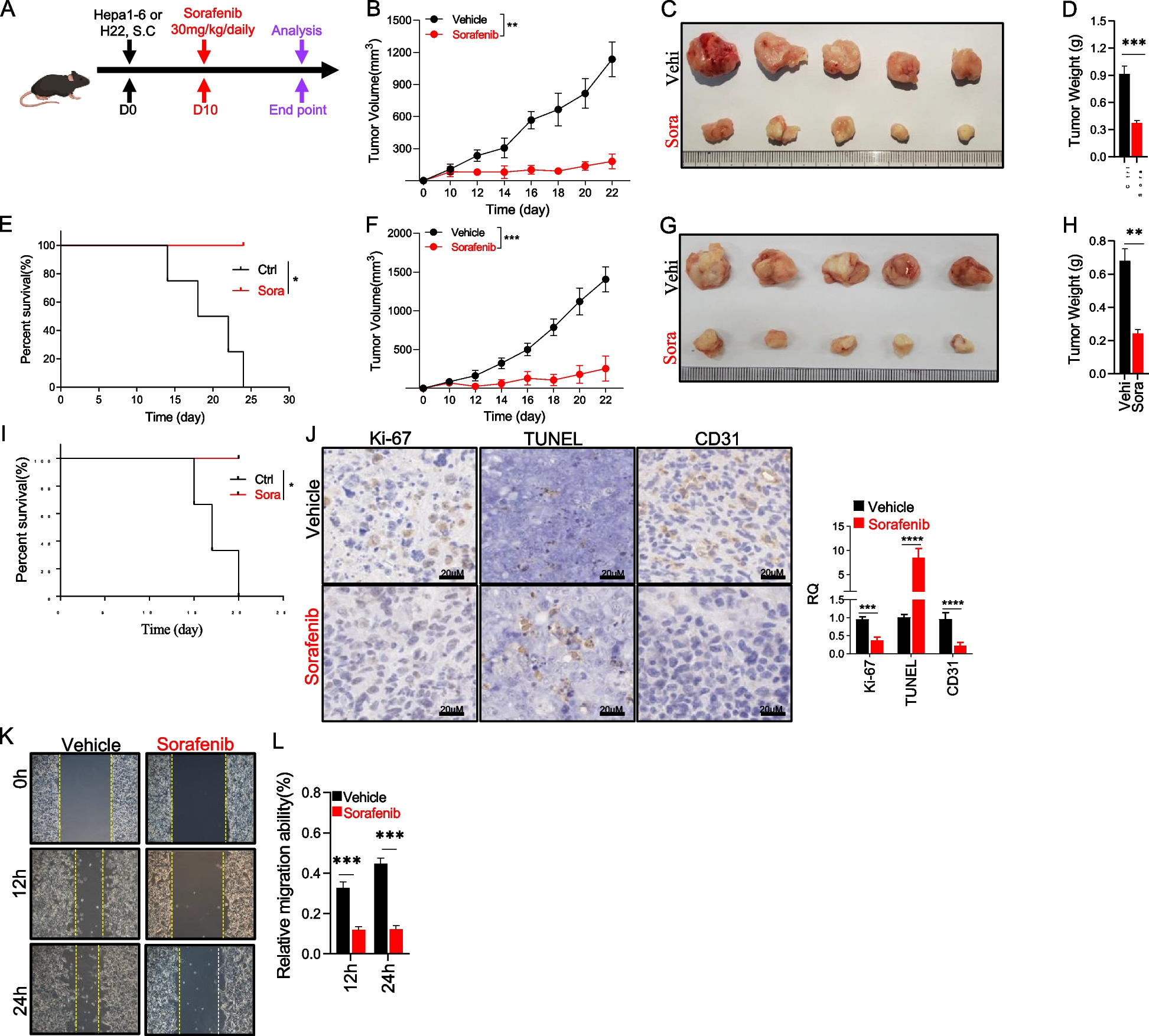
A mouse model of xenograft breast tumors was established, as previously described [21]. Female BALB/c nude mice were injected with 106 SCP2/WT, SCP2/NC or SCP2/KD cells via the left ventricle (n = 8–10 per group) and their body weights were monitored for 42 days after inoculation. The bone metastatic colonization of breast tumor cells was monitored by X-rays and bioluminescence weekly, and the survival of tumor-bearing mice was checked daily.
For the drug treatment assay in vivo, individual mice receiving any type of SCP2 cells were randomized and treated intravenously with vehicle (5% of DMSO in PBS) or 10 mg/kg S6 twice per week for 42 days (n = 8–10 per group). Individual tumor cell-bearing mice were monitored longitudinally, and their body weights were measured every other day. The mice were anesthetized and euthanized when the mice lost 20–25% of their original body weights or developed symptoms of cachexia or wasting. The tumors in the bone and vertebrae were dissected and microCT photographed, followed by fixation, decalcification, embedding and H&E staining. The tumor tissue sections were stained with Tartrate-resistant acid phosphatase (TRAP) using a specific kit (387A, Sigma). Their heart, liver, spleen, lung, bone marrow and kidney tissues were collected, sectioned, and stained with H&E to observe potential S6 toxicity. The animal experiments were carried, per the Guide for the Care and Use of Laboratory Animals of National Institutes of Health Guide and the protocols were approved by the Institutional Research Ethics Committee of Shengjing Hospital of China Medical University (Project ID 2020PS318K, approved on 04/01/2020).
Molecular modeling and docking
The intact structure of the major domains (A135-L731) of STAT3 was established, based on the crystal structures (Protein Data Bank [PDB] code 6QHD) and Alphafold2 model of STAT3 (UniProt ID P40763) using Modeller 9.22 with 1,000 decoys. The structure of STAT3 was evaluated using DOPE, Molpdf score, DFIRE2, and Procheck, followed by 50-ns molecular dynamics (MD) relaxation.
The loop1 and loop3 of the SOST NMR structure (PDB code 2K8P) of two beta-sheet structures of SOST protein were selected with the relaxed STAT3 modeling structure, residues 75–81 and 132–138, respectively. Rigid-body docking for SOST with STAT3 was performed with Zdock program to obtain the potential protein–protein interaction poses, with some key residues preferred during the docking process. The complex with the best docking score was used for 100-ns MD optimization and relaxation.
All MD simulations were performed using Gromacs 2020.4 with Amber14 force field. The structure was solvated in a cubic TIP3P water box with 1 nm distance from the edge, which was neutralized by adding appropriate number of sodium and chloride ions. After two steps of energy minimization, this system was gradually heated to 300 ºK over 100 ps to perform the 2-ns NVT equilibration and 5-ns NPT equilibration. Finally, MD simulations at 300 ºK and 1 atm were carried out with the LINCS algorithm to restrain hydrogen positions at their equilibrium distances, which allowed the use of an integration time step of 2 fs. Both energies and coordinates were saved every 10 ps for postproduction analysis. All simulations were performed on a high-performance computer cluster running the Linux operating system. The MM-PBSA calculation was conducted using the gmx_MMPBSA tool [22].
Virtual screening
The final frame of the modeling structure from the MD simulation was used for site prediction and virtual screening. FTSite [23] and FTMap [24] were used to detect the binding pocket of SOST. Our in-house docking program, FIPSDock [25], was used to screen a library against SOST. The geometric center of the SOST structure was chosen as the grid center. Each grid contained 200 × 200 × 160 grid points with 0.375 Å spacing to cover the whole SOST structure. To narrow down the virtual screening results effectively, we chose only the best docked structures with the lowest binding free energies for each possible binding mode to estimate the binding pose and important interactions. Notably, the S6 molecule stood out among the top 200 hits. The complexed structure of SOST with the best pose of S6 was built to conduct 500-ns MD simulation.
Testing drug sensitivity in organoid cultures
Surgical breast tissues were obtained from a patient with breast cancer at Shengjing Hospital of China Medical University after the patient provided informed consent. The tissues were minced using a scalpel, cultured into a 50-mL C-tube containing 20 mL of AdDF + + + (Advanced DMEM/F12 with 1 × Glutamax, 10 mM HEPES, and 1% P/S) and 2 mg/mL collagenase at 37 °C for 2 h with general shaking. After digestion, the suspension was centrifuged at 400 g for 10 min; mechanically sheared by pipetting with 10, 5, and 1 mL pipette tips; and resuspended in 10 mL of AdDF + + + containing 2% of FBS each time to obtain organoids. The cell suspension was added to Matrigel at a 1:2 ratio; 100 µL of the solution was then placed at the center of each well of a 24-well culture plate; the gel was solidified at 37 °C for 30 min in 5% CO2. The medium was replaced every 3 days and organoids were passaged weekly. The growth of organoids was evaluated under an inverted microscope, and the organoids with fine growth conditions that had been more than 3 passages were used for subsequent experiments. The organoids were treated with, or without (NS), 2 μM S6, or 80 μM S6 for 48 h and the potential toxicity was evaluated under an inverted microscope (Nikon, Japan).
Differentiation of bone marrow mesenchymal stem cells (BMSCs)
To induce osteoblast differentiation, the isolated mouse BMSCs (2 × 105 cells/well) were treated with DMSO or S6 (8 µM) in a modified α-MEM containing 10% of FBS, 50 μg/ml L-ascorbic acid, 1 mM dexamethasone and 1 M β-glycerophosphate in 12-well plates. The media were changed every other day for 14 days. The cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% of Triton-X 100, denatured again with 0.1% PBST, and stained with alkaline phosphatase (ALP) using a specific kit (Sigma-Aldrich, USA). Some cells were cultured for 21 days and stained with alizarin red staining (ARS). Subsequently, the culture plates were washed with distilled water and treated with 10% of cetylpyridinium chloride for 2 h with general shaking to fully dissolve the mineralized nodules. The relative absorbance value of S6 to the control (DMSO) solution (designated as 1) was evaluated and calculated.
Transcriptome sequencing and promoter prediction
SCP2/NC or SCP2/KD1 cells were harvested, and their total RNA was extracted, followed by transcriptome sequencing on illumina platform (Biomarker Technologies, Beijing, China). The levels of original gene mRNA transcripts were analyzed using the FPKM method and DESeq2. The differentially expressed genes (DEGs) were defined when log2FC > 1 or < − 1, and false detection rate (FDR) < 0.05. The potential pathways of DEGs were analyzed by KEGG pathway annotation. The STAT3 binding sites in the TGF-β and KRAS promoter regions were predicted using rVista 2.0 software in JASPAR database [26].
Statistical analysis
Data are expressed as means ± standard error of mean (SEM). Differences among groups were analyzed by one-way analysis of variance (ANOVA) and post hoc Newman-Keuls test. Differences between groups were analyzed by Student’s t-test. DMFS in each group of patients was estimated using the Kaplan–Meier method and analyzed by the log-rank test. Risk factors associated with poor survival were identified using univariate and multivariate regression analyses with the Cox regression model. All statistical analyses were performed using SPSS 23.0 software (IBM Corps., Armonk, NY, USA). Statistical significance was set at a P-value of < 0.05.





留言 (0)