
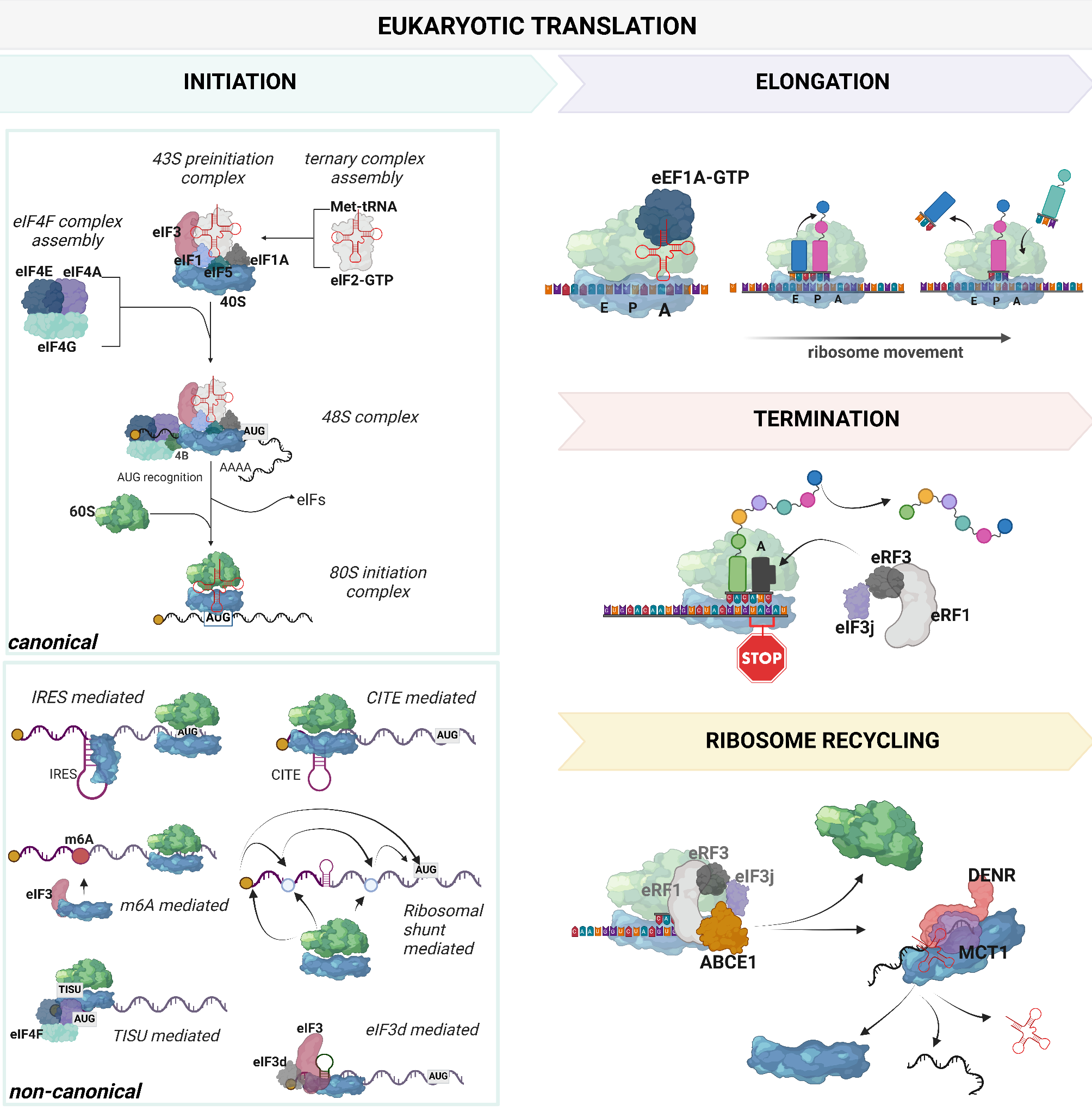
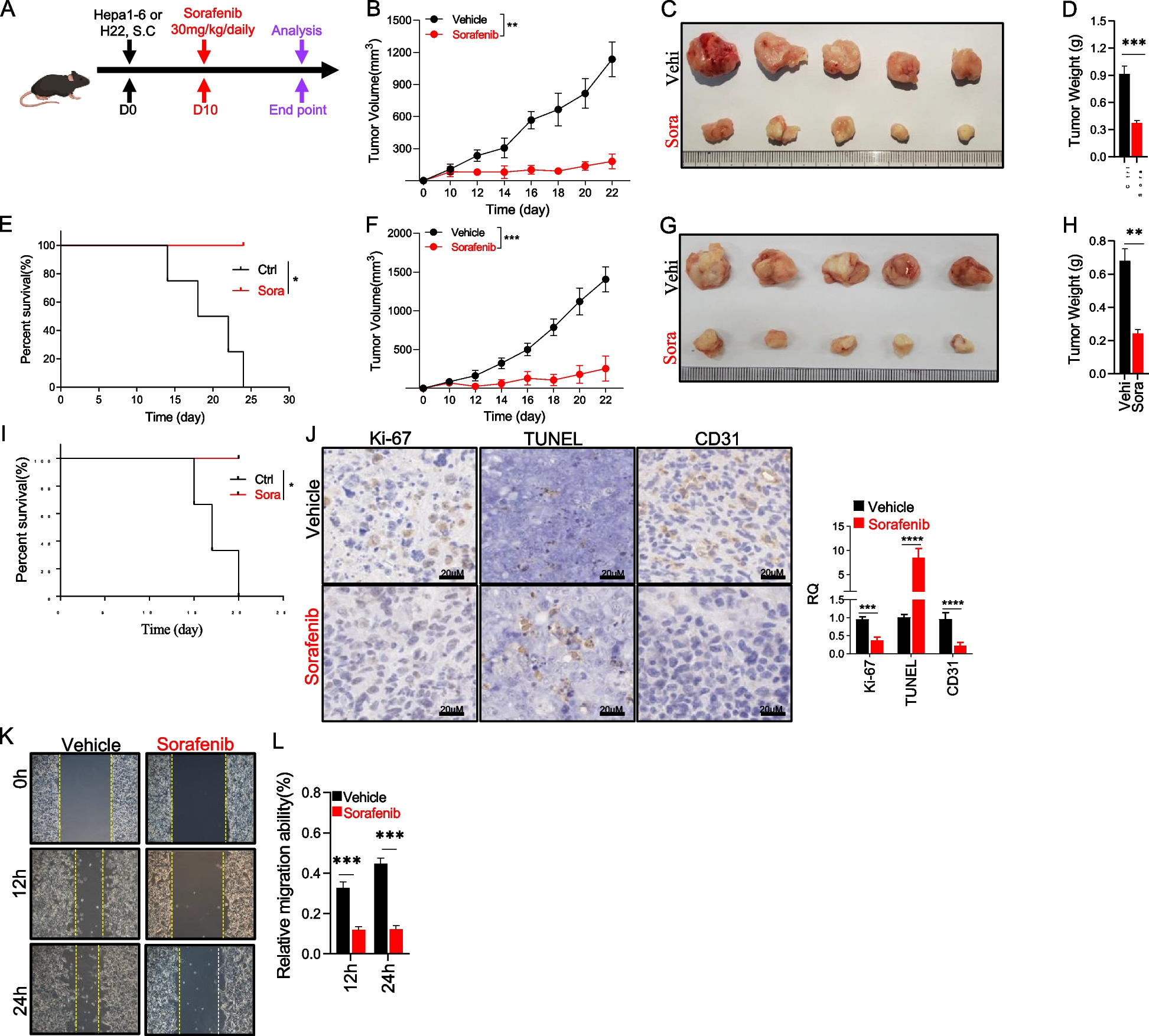
記住我




Alendronate-hyaluronic acid graft polymer coated quercetin-loaded manganese phosphate nanoparticles (AHA@MnP/QCT NPs) were synthesized following a previously reported method [27]. The process involved three steps: (i) Activation of the carboxylic groups on HA by adding N-hydroxysuccinimide (NHS) and 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), which enabled grafting with alendronate to form alendronate-hyaluronic acid graft polymer (AHA). (ii) A stable core was then formed by mixing MnCl2 with QCT through a complexation reaction. (iii) 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer containing AHA was added dropwise to the above mixture under ultrasonic conditions. As shown in Scheme 1, the phosphate group in AHA encapsulates the Mn2+-QCT complex, forming negatively charged AHA@MnP/QCT NPs. Various concentrations of Mn2+ and AHA were investigated to determine their influence on particle size, polydispersity index (PDI), zeta potential and QCT encapsulation efficiency (Table S1-2). Additionally, cellular uptake of different nanoparticles by A549 cells was quantified using flow cytometry (Figure S1). Finally, the optimal formulation was determined to be 100 mM Mn2+ and 10 mg/ml AHA for nanoparticle preparation.
Scheme 1
Schema of the preparation of AHA@MnP/QCT nanoparticles and its one-stone-for-two-birds strategy to attain the integrated diagnosis and treatment of non-small cell lung cancer. The AHA@MnP/QCT NPs vector sustainably releases QCT and Mn2+ into the acidic environment, which induces apoptosis and promotes ferroptosis in cells via the Fenton-like reactions. Free Mn2+ induces immunogenic cell death by activating DCs and promoting the activation and proliferation of T cells. Non-invasive imaging is achieved by accumulating AHA@MnP/QCT and enhancing T2-MRI signal at the tumor site
Following mineralization, the AHA@MnP/QCT NPs exhibited a translucent yellowish-brown appearance without noticeable precipitation (Fig. 1A). In contrast, MnP NPs lacking AHA encapsulation was unsuitable as carriers due to their propensity for aggregation and precipitation, a result of infinite core growth (Figure S2 and S3). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images showed that the AHA@MnP/QCT NPs were monodispersed, displaying a well-defined spherical morphology with a smooth surface (Fig. 1B-C). The mean diameter of the NPs was measured to be between 120 and 150 nm. For further characterization of synthesized NPs, elemental analysis using energy-dispersive spectroscopy (EDS) confirmed the presence of C (24.5%), O (44.5%), Mn (24.7%) and P (6.3%) on the NPs surface, indicating successful nucleation and growth of MnP/QCT minerals within the AHA matrix (Fig. 1D-E). Dynamic light scattering (DLS) analysis demonstrated a hydrodynamic diameter of 205.60 nm with a polydispersity index (PDI) of 0.20 (Fig. 1F). The AHA@MnP/QCT NPs showed good homogeneity compared to the hydrated particle size of each of the other components (Figure S4). The zeta potential measurements indicated distinct charges for each component: -14.03 mV (QCT), + 2.47 mV (MnCl2), -2.34 mV (MnP NPs), -4.24 mV (AHA@MnP NPs) and − 18.6 mV (AHA@MnP/QCT NPs) (Fig. 1G). The slightly larger particle size measured by DLS likely results from the Brownian motion of the NPs in solution and the presence of a few solvent layers [28]. Fourier transform infrared spectrometry (FTIR) data (Fig. 1H) showed strong characteristic peaks at 872, 1043, 1171, 1460, and 3404 cm− 1, corresponding to the vibration of different characteristic functional groups within MnP NPs. The fluctuations at 872 and 1043 cm− 1 may be related to the vibration of manganese oxides, 1171 and 1461 cm− 1 are characteristic absorption peaks of phosphate, and the characteristic peak at 3404 cm− 1 is considered to be a hydroxyl-related peak. AHA@MnP NPs and AHA@MnP/QCT NPs, and the characteristic peaks were attributed to. The diminished intensity of these absorption peaks in AHA@MnP NPs and AHA@MnP/QCT NPs compared to MnP NPs suggested the encapsulation of AHA and the complexation of QCT, which lowers the concentration of MnP at equivalent mass, thereby indicating the successful integration of AHA, QCT and MnP. X-ray photoelectron spectroscopy (XPS) further confirmed NPs formulation, with binding energy of Mn 2p at 641.08 eV, O 1s at 531.08 eV, C 1s at 285.08 eV and P 2p at 168.08 eV for AHA@MnP/QCT NPs (Fig. 1I). The addition of AHA and QCT increased the intensity of P 2p and O 1s relative to MnCl2, MnP NPs and AHA@MnP NPs (Figure S5). Further peak splitting of the XPS spectrum of Mn 2p revealed that three distinct peaks corresponding to Mn2+ (54.94%), Mn3+ (27.47%), and Mn4+ (17.59%) with binding energies of 640.98 eV, 643.33 eV and 646.48 eV, respectively (Fig. 1J). Similarly, the O 1s spectra showed peaks at 530.98 eV, 531.78 eV, and 536.28 eV, attributed to P-O, metal-oxygen bonding (Mn-O-Mn), and surface oxygen (water, O = C-O), respectively (Figure S6). The Ultraviolet-visible (UV − vis) spectrophotometry confirmed characteristic absorption peaks: MnP NPs and AHA@MnP NPs exhibited a distinct peak at 206 nm, indicative of MnP core formation, while QCT and AHA@MnP/QCT NPs displayed a typical peak at 300 nm, confirming the presence of QCT (Fig. 1K). Based on the absorbance at 300 nm, the content of QCT in AHA@MnP/QCT NPs was calculated to be 12.3% with an encapsulation efficiency of 93.2% (Table S2). Collectively, these results demonstrated the successful synthesis of AHA@MnP/QCT NPs.
Fig. 1
Characterization of AHA@MnP/QCT NPs. (A) The pictures of QCT, AHA@MnP and AHA@MnP/QCT; (B-C) Representative SEM (B) and TEM (C) image of AHA@MnP/QCT NPs; (D) EDS mapping characterization images of AHA@MnP/QCT NPs; (E) EDS characterization of AHA@MnP/QCT NPs; (F) Hydrodynamic size distribution of AHA@MnP/QCT NPs; (G) Zeta potential of QCT, MnCl2, MnP, AHA@MnP and AHA@MnP/QCT NPs; (H) FT-IR spectra of AHA@MnP/QCT, AHA@MnP and MnP NPs; (I) XPS analysis of AHA@MnP/QCT NPs; (J) The peaks of Mn 2p of AHA@MnP/QCT NPs; (K) UV − vis spectra of nanoparticles
The stability of the AHA@MnP/QCT NPs was assessed through various metrics, including dilution stability, media compatibility, and long-term storage by determining the particle size of the NPs. Remarkably, The NPs retained their particle size (~ 270 nm) even after 512-fold dilution and the size remained (Figure S7A) consistent across different media, indicating robust dilution stability and the adaptability of these NPs in diverse biological environments (Figure S7B). Notably, a slight increase in particle size was observed in phosphate buffered saline (PBS) solution, likely due to the elevated phosphate concentration disrupting the AHA encapsulation and promoting growth of the MnP core. Moreover, the NPs exhibited long-term stability with minimal changes in size when stored at 4 °C for up to three weeks (Figure S7C). In summary, these findings indicated the exceptional stability of AHA@MnP/QCT NPs, positioning them as promising candidates for in vivo therapeutic applications.
Cellular uptake and release mechanisms of AHA@MnP/QCT NPsThe cellular uptake of NPs is a critical determinant of pharmacological efficacy, influencing their intracellular fate, biological response, and overall therapeutic potential within tumor environments [29]. Biocompatible hyaluronic acid (HA) is often chosen as both a “target head” and “stabilizer”, ensuring the stability of the NPs and improving tumor targeting [30]. The incorporation of HA endowed the NPs with a strong negative charge and facilitated specific targeting of the CD44 receptors on tumor cells [31], which is overexpressed in non-small cell lung cancer cells. The fluorescence intensity of FITC labeled AHA@MnP/QCT NPs in Lewis cells was quantified by flow cytometry, and it was slightly lower than that of the Lipo2000/FITC, but significantly higher than that of all other groups (Fig. 2A). After a 6 h incubation, the fluorescence intensity of AHA@MnP/FITC NPs was approximately four times greater than MnP/FITC NPs and ten times greater than free FITC (Fig. 2B). In addition, pre-treatment with HA significantly reduced the fluorescence intensity of AHA@MnP/FITC NPs, which was not significantly different from free FITC, supporting the critical role of HA in CD44 receptor-mediated targeting during NPs uptake. Similar trends were observed in laser confocal microscopy images, where AHA@MnP/QCT NPs and Lipo2000/Cy5 showed strong enrichment in tumor cells with more Cy5 (red)-labeled fluorescence (Fig. 2C). Similarly, the uptake of AHA@MnP/FITC NPs in A549 cells was six times higher than MnP/FITC NPs and 13 times higher than free FITC by measuring intracellular fluorescence intensities using flow cytometry (Figure S8A-B). The strong fluorescence of AHA@MnP/QCT NPs and the weak fluorescence of HA pretreatment in confocal images also further illustrate the important role of HA in targeting towards CD44 receptor in tumor cells by NPs (Figure S8C).
The release behavior of AHA@MnP/QCT NPs was further investigated under varying pH conditions to clarify their sensitivity to the acidic tumor microenvironment. Upon mixing the AHA@MnP/QCT NPs with HEPES buffer solutions at different pH levels (pH 7.4, 6.5, 5.0) and then centrifuging, black precipitates appeared at pH 7.4 and 6.5, while a translucent solution stated at pH 5.0 (Fig. 2D). This trend was paralleled by a gradual increase in particle size and PDI as the pH decreased, indicating slow dissolution of the NPs at lower pH values (Fig. 2E). SEM images further corroborated this finding, showing the transformation of smooth spherical particles into irregular scales as pH shifted from 7.4 to 5.0. (Fig. 2F). Notably, Mn2+ and QCT release profiles confirmed the pH sensitivity of the NPs. Absorbance values at 525 nm were determined by potassium periodate method to quantify the release of Mn2+ from NPs in HEPES buffer solutions at different pH. After 2 h of release, the release rate of Mn2+ was only 18.18% at pH 7.4, but increased sharply to 87.12% at pH 5.0 (Fig. 2G). Similarly, QCT release rates quantified at 300 nm were 8.77%, 12.13%, and 84.89% at pH 7.4, 6.5, and 5.0, respectively (Fig. 2H). In summary, these results showed the ability of AHA@MnP/QCT NPs to remain stable under physiological conditions (pH 7.4), but rapidly disintegrate within acidic tumor environment, facilitating localized drug release.
The intracellular release of NPs within the Lewis cells was visualized using laser confocal microscopy, showing that Cy5 fluorescence (the red dots) gradually escaped from lysosome markers (the green dots) over time, indicating that the colocalization of Cy5 with the lysosome gradually weakened (Fig. 2I). After 6 h, the colocalization coefficient of Cy5 and lysosomes decreased from 85.7 ± 4.00% at 2 h to 33.8 ± 10.74% (Figure S9), indicating efficient lysosomal escape. The color colocalization analysis of the cell sections (shown by the white line) further illustrated the NPs labelled by Cy5 (red line segments) effectively released from lysosomes (green line segments) from 2 h to 6 h (Fig. 2J-K). Similar assays were conducted on A549 cells for clinical applications. The colocalization coefficients of Cy5 and lysosomes were also time-dependent from 87.7 ± 2.95% at 2 h to 47.9 ± 10.09% at 8 h, showing a slower release profile (Figure S10A-B). The slower release of NPs in A549 cells compared to Lewis cells suggested that AHA@MnP/QCT NPs may have sustained anti-tumor activity, contributing to prolonged therapeutic effects (Figure S10C-D).
In conclusion, the cellular uptake and release mechanisms of AHA@MnP/QCT NPs in tumor cells were illustrated in Fig. 2L. Upon proximity (Step 1), signal recognition (Step 2), and uptake via HA-mediated CD44 receptor interaction (Step 3), the NPs dissolve under acidic lysosomal conditions (Step 4), triggering rapid osmotic pressure increase that lead to lysosomal rupture and intracellular drug release from NPs. The intracellular and extracellular processes of NPs as shown in red boxes (Fig. 2M-N) were also confirmed by TEM images of AHA@MnP/QCT NPs-treated Lewis cells, corresponding with the schematic illustration. Compared to untreated cells, AHA@MnP/QCT NPs-treated cells had more black particles and rougher cell edges, which was attributed to the accumulation of incompletely disintegrated NPs inside the cells and their continued pharmacological effects causing tumor cell death. These findings provide critical insights into the mechanism of action of AHA@MnP/QCT NPs and their potential for targeted cancer therapy.
Fig. 2
The endocellular mechanisms and controlled realease of AHA@MnP/QCT NPs. (A-B) Quantitative analysis of the Lewis cells intracellular fluorescence intensities by flow cytometry (FITC: 10 µg/mL, n = 3); (C) The Lewis cells intracellular fluorescence intensities detected by confocal microscopy; (D) Disslution of AHA@MnP/QCT NPs in different pH solutions; (E) The changes of particle size (blue) and PDI (red) for different pH HBS solutions; (F) SEM of AHA@MnP/QCT NPs in different pH HBS solutions; (G-H) Mn2+ (G) and QCT (H) release from AHA@MnP/QCT NPs in HBS with different pH values; (I) Confocal imaging of co-localization of AHA@MnP/QCT NPs (Cy5-labeled, red) with lysosomes (Lyso Tracker green, green) in Lewis cells. Hoechst 33,258 (blue) was used for staining the nucleus (Scale bar: 10 μm); (J-K) Fluorescence intensity of Lewis cells section at 2.0 (J) and 6.0 (K) hours; (L) The schematic illustration of transmembrance uptake and lysosomal escape of AHA@MnP/QCT NPs; (M-N) TEM imaging of cellular morphology with untreatment (M) and treatment (N) of AHA@MnP/QCT NPs
The anti-tumor effect of AHA@MnP/QCT NPs in vitroTo explore the therapeutic effects of AHA@MnP/QCT NPs against cancer cells, we employed multiple assays, including the CCK-8 cell counting kit, Annexin V-FITC/PI flow cytometry and Western blotting assay. As shown in Fig. 3A and S11A, severe inhibition of both cancer cell lines (Lewis and A549 cells) was observed after 48 h of treatment with QCT and AHA@MnP/QCT NPs. This inhibitory effect was concentration-dependent, with QCT-treated cells showing approximately 40% cell death at a concentration of 10 µg/mL, while the AHA@MnP/QCT NPs-treated group exhibited 60% cell death at just 5 µg/mL. These findings suggested that not only was QCT effective in suppressing tumor growth, but the NPs significantly enhanced its therapeutic potential, possibly due to the synergistic effects of Mn2+ and QCT. The AHA@MnP NPs alone also induced about 50% tumor cell death at 5 µg/mL (Fig. 3B and S11B), further showing their standalone therapeutic effect without QCT. As shown in Fig. 3C, numerous apoptotic vesicles were present in Lewis cells following AHA@MnP/QCT NPs treatment, suggesting that NPs induced apoptosis in tumor cells. In addition, substantial autophagic vesicles were also observed, implying that NPs elicited their effects through multiple pathways rather than inducing apoptosis alone (Figure S12). Flow cytometry assays corroborated these findings, demonstrating that AHA@MnP/QCT NPs significantly enhanced the therapeutic effects on both Lewis and A549 cells compared to QCT alone and AHA@MnP treatment (Fig. 3D and S13). Specifically, AHA@MnP/QCT NPs increased tumor cell death by 5.5-fold in Lewis cells and 4-fold in A549 cells relative to the single action of QCT (Fig. 3E and S14). Similarly, the apoptotic status of Lewis cells was observed by using the method of PI/Calcein-AM double staining, and similar results were obtained (Fig. 3F). To delve deeper into the underlying mechanisms of enhanced tumor cell death, we investigated the expression of proteins from Bcl-2 family and the caspase cascade reactions, which are pivotal regulators of apoptosis. Western blot analysis (Fig. 3G and S15) revealed that treatment with QCT, AHA@MnP and AHA@MnP/QCT NPs led to upregulation of the pro-apoptotic protein Bax. In contrast, the expression of the anti-apoptotic protein Bcl-2 and the inactive effector protein P-Caspase-3 was downregulated, suggesting that the treatment shifts the balance toward apoptosis [32, 33]. These results indicated that AHA@MnP/QCT NPs induced apoptosis through the intrinsic mitochondrial pathway, further enhancing the cytotoxic effects of QCT.
Fig. 3
In vitro treatment effects of AHA@MnP/QCT NPs. (A-B) Relative viabilities of Lewis cells aftertreatment with different concentrations of QCT and nanoplexes for 48 h (n = 6); (C) TEM imaging of the formation of apoptotic bodies with aftertreatment of AHA@MnP/QCT NPs (red arrows: NPs uptake; yellow box: apoptotic vesicle exocytosis); (D) Flow cytometry analysis of Annexin V-FITC/PI costained Lewis cells; (E) Quantitative analysis the apoptosis rate of Lewis cells; (F) PI/Calcein-AM double staining images of Lewis cells; (G) The expression of P-Caspase-3, Bcl-2 and Bax protein for Lewis cells treated with QCT, AHA@MnP or AHA@MnP/QCT NPs
In conclusion, these findings confirmed the potent anti-tumor activity of AHA@MnP/QCT NPs, driven by their ability to enhance apoptosis via Bax/Bcl-2 regulation and activation of the caspase cascade. The results showed the potential of these NPs not only to potentiate the effects of QCT but also to serve as a multifaceted therapeutic strategy that simultaneously targets apoptotic and autophagic pathways.
Pharmacodynamic mechanism of AHA@MnP/QCT NPsTo further determine the anti-tumor pharmacodynamic mechanism of AHA@MnP/QCT NPs, the gene transcriptomic analysis on Lewis cells was conducted by treating with PBS (labeled: Ctrl), AHA@MnP (labeled: AM), and AHA@MnP/QCT (labeled: AMQ). Initially, differentially expressed genes (DEGs) were identified between the AM versus Ctrl and AMQ versus Ctrl groups, revealing 1537 and 1649 DEGs, respectively (Fig. 4A). Compared with the Ctrl group, the DEGs exhibited distinct regulation patterns, with up-regulated genes (red) in the AM and AMQ groups primarily associated with immune effector processes, tumor necrosis factor (TNF) production, apoptotic signaling pathways, and reactive oxygen species (ROS) response (Fig. 4B). In contrast, down-regulated genes (blue) were linked to glycolytic processes, epithelial cell differentiation, ATP metabolism, cell adhesion, and cell development (Fig. 4C). These findings suggested that AHA@MnP/QCT NPs exerted their medicinal effects by promoting tumor cell death and disrupting their metabolic processes through multiple signaling pathways [34].
To further detect the role of ROS in tumor cell death induced by NPs, we analyzed the expression of ROS-related genes in each group (Fig. 4D). Notably, both AM and AMQ groups showed reduced expression of ROS-related genes such as Glrx2 and Sod1 compared with the Ctrl group [35, 36]. The similarity in ROS gene expression between the AMQ and AM groups may be attributed to their equivalent Mn2+ concentrations, highlighting the important role of Mn2+ in driving ROS-mediated pathways (Fig. 4E). The involvement of TNF signaling, necrosis, and apoptosis pathways was confirmed through Gene Set Enrichment Analysis (GSEA) (Fig. 4F). Quantitative analysis of necrotic and apoptotic pathways up-regulation revealed that both AM and AMQ significantly increased these signals, with AMQ showing the highest percentage of up-regulation (Fig. 4G). This suggested that tumor cell death induced by AHA@MnP/QCT NPs operates through multiple pathways, where both QCT and Mn2+ play important roles in mediating the cytotoxic effects, and Fig. 3D and Figure S12 also provide support for this conclusion.
In summary, AHA@MnP/QCT NPs exert their therapeutic efficacy mainly through three interconnected pathways: (i) Increasing the accumulation of ROS, causing mitochondrial and endoplasmic reticulum rupture, and inducing the ferroptosis of tumor cells; (ii) Up-regulating apoptosis and necroptosis pathways through multiple pathways, which contributes to the production of apoptotic and autophagic vesicles; (iii) Stimulating of immune response, which induces the organism to carry out the immune killing of tumor cells.
Fig. 4
Pharmacodynamic mechanism of AHA@MnP/QCT NPs. (A) Volcano plots showing DEGs between AHA@MnP NPs (labeled: AM) and Control (labeled: Ctrl), AHA@MnP/QCT NPs (labeled: AMQ) and Ctrl; (B) Heatmap depicting the enrichment of upregulated genes between AM and Ctrl, AMQ and Ctrl; (C) Heatmap depicting the enrichment of down-regulated genes between AM and Ctrl, AMQ and Ctrl; (D) Box plot depicting the enrichment of Ctrl, AM, and AMQ in ROS-related genes; (E) Heatmap depicting the expression levels of Ctrl, AM, and AMQ in ROS-related genes; (F) The GESA plot showed that the TNF signaling pathway, necroptosis, and apoptosis pathways were up-regulated in the AM and AMQ group compared with the Ctrl group; (G) Bar graph depicting the proportion of necroptosis and apoptotic pathway upregulated in AM and AMQ group
Multiple targeted therapy - FerroptosisMn2+, released under acidic conditions, contribute to ferroptosis in tumor cells by interacting with H2O2 to initiate a Fenton-like reaction, causing mitochondrial and endoplasmic reticulum rupture (Fig. 5A). To investigate the anti-tumor mechanism of AHA@MnP/QCT NPs, we examined the effects of NPs on H2O2, GSH/GSSG, and ROS both in vitro and in tumor cell models. In vitro, the decomposition of H2O2 by AHA@MnP/QCT NPs was determined by adding the NPs to a PBS solution containing 100 µM H2O2. The appearance of bubbles, highlighted by red boxes, indicated the generation of oxygen (O2), signaling the breakdown of H2O2 (Fig. 5B). Quantitative analysis using a hydrogen peroxide analysis kit revealed that H2O2 was nearly fully decomposed within 3 h (Fig. 5C). The proposed mechanism involved the release of Mn2+ through the following reactions: (i) Mn3(PO4)2 (s) + 4 H+ → 3Mn2+ + 2H2PO4−. (ii) Mn2+ + H2O2 → MnO2 + 2 H+. (iii) MnO2 + H2O2 + 2 H+ → Mn2+ + 2H2O + O2↑ [37]. As shown in Figure S16, treatment with different NPs resulted in a significant downregulation of Eno1 expression, suggesting a marked reduction in intracellular glycolysis. This metabolic shift likely increased the availability of oxygen within the tumor microenvironment, while concurrently enhancing the consumption of hydrogen peroxide [38]. Furthermore, Mn2+ promoted the Fenton-like reactions (Mn2+ + H2O2 + H+ → Mn3+ + H2O + ·OH), which converted GSH to GSSG, contributing to oxidative stress. To assess ·OH production in an acidic environment (pH ~ 4), we used methylene blue (MB) as an indicator. MB degrades in the presence of ·OH, leading to decreased absorbance [39]. While H2O2 alone caused minimal absorbance change (~ 4%), AHA@MnP/QCT NPs led to a ~ 50% decrease, likely due to the partial influence of NPs on the absorbance measurement of MB. The combination of AHA@MnP/QCT NPs and H2O2, however, resulted in a significant decrease (~ 65%), confirming the NPs’ ability to generate ·OH efficiently via a Mn2+-mediated Fenton-like reaction (Figure S17). In tumor cell models, we quantified H2O2 and GSSG levels in NP-treated Lewis cells using a hydrogen peroxide kit and a GSH-GSSG kit. NPs caused a statistically significant decrease in H2O2 (by 50%) and a statistically significant increase in GSSG (by 125%), compared with untreated Lewis cells (Fig. 5D-E). in A549 cells, similar effects were observed, with a 75% decrease in H2O2 and a 315% increase in GSSG (Figure S18).
Images of TEM showed that unlike the smooth mitochondrial surface in untreated Lewis cells, mitochondria in Lewis cells treated with AHA@MnP/QCT NPs for 6 h appeared hollow and ruptured, with fragmented endoplasmic reticulum (Fig. 5F). This damage was attributed to excessive accumulation of ROS, which triggered ferroptosis. As shown in Figure S19A-B, a large accumulation of ROS happened after 2 h of AHA@MnP/QCT NPs treatment in Lewis cells, implying the rapid action of NPs. Additionally, flow cytometry confirmed that ROS levels significantly increased in Lewis cells after 2 h of AHA@MnP/QCT NPs treatment, reaching 2.0-fold higher levels compared to untreated cells (negative control) and 1.5-fold higher than the ROSup positive control (Fig. 5G). Laser confocal microscopy further validated these results, showing where more fluorescent green dots (DCFH-DA, a probe for quantitative detection of ROS) in AHA@MnP/QCT NPs treated Lewis cells, suggesting that the NPs promoted ROS production (Fig. 5H). In A549 cells, ROS production followed a time-dependent pattern, peaking after 6 h of NPs treatment and leveling off by 8 h (Figure S19C-D). After 8 h, ROS levels in AHA@MnP/QCT NPs-treated cells were 2.9 higher than untreated cells and 1.8-fold higher than the ROSup group, with only a slight difference from the AHA@MnP NPs group (1.2-fold higher) (Figure S19E-F). This was supported by the fluorescence profile of the laser confocal microscope (Figure S19G). Accumulation of reactive oxygen species (ROS) can induce lipid peroxidation within cells, subsequently leading to cellular damage. To validate the peroxidation status in tumor cells, the BODIPY 581/591 C11 probe and Western blot analysis were employed. The results demonstrated that in the AHA@MnP NPs, AHA@MnP/QCT NPs, and LPOup groups, a higher proportion of oxidized BODIPY was detected, while the control group predominantly exhibited the reduced state (Fig. 5I-J) [40]. Concurrently, the Western blot results also indicated a significant upregulation of lipid peroxidation (LPO) protein expression in tumor cells following nanoparticle (NPs) treatment (Fig. 5K). These results suggested that AHA@MnP/QCT NPs can efficiently produce ·OH via a Fenton-like reaction, causing a decrease in intracellular reducing substances and an increase in free radicals. This led to excessive ROS accumulation in mitochondria and the endoplasmic reticulum, ultimately inducing ferroptosis in tumor cells [41].
Fig. 5
Mechanism of ferroptosis generation in AHA@MnP/QCT NPs. (A) Schematic illustration for AHA@MnP/QCT NPs involved in the ferroptosis process; (B) Image of H2O2 decomposition after treatment with different nanoplexes; red farme, gas bubble; (C) curve of H2O2 decomposition (n = 3); (D) Intracellular H2O2 level for Lewis cells treated with AHA@MnP NPs or AHA@MnP/QCT NPs; (E) Intracellular GSSG level for Lewis cells treated with AHA@MnP NPs or AHA@MnP/QCT NPs; (F) Tem images of mitochondria (a-b) and endoplasmic reticulum (c) in Lewis cells before and after AHA@MnP/QCT NPs treatment; (G) Quantitative analysis of the Lewis cells intracellular fluorescence intensities after treatment with different nanoplexes by flow cytometry; (H) Confocal images of DCHF-DA-stained Lewis cells pre-treated AHA@MnP NPs or AHA@MnP/QCT NPs (Scale bar: 100 μm); (I) Confocal images of BODIPY 581/591 C11-stained Lewis cells pre-treated AHA@MnP NPs or AHA@MnP/QCT NPs (Scale bar: 50 μm); (J) Quantitative analysis of the Lewis cells intracellular fluorescence intensities after treatment with different nanoplexes (n = 3); (K) The expression of LPO protein for Lewis cells treated with AHA@MnP NPs or AHA@MnP/QCT NPs. ****P < 0.001, ***P < 0.001
Multiple targeted therapy - immunogenic cell deathMn2+ has been shown to cause immunogenic death of tumor cells through activation of the cGAS-STING pathway [42]. To explore this ICD mechanism, we conducted a series of protein blotting assays to assess key ICD biomarkers. As shown in Fig. 6A-B, treatment with various NP formulations led to a marked upregulation of cGAS and phos-STING/STING, indicative of ICD activation and subsequent engagement of the cGAS-STING signaling axis. This activation is crucial for the proliferation and maturation of dendritic cells and the subsequent activation of T cells [43]. To further validate the induction of ICD, we measured the release of ATP, HMGB1 and calreticulin (CRT) using both commercial assay kits and immunofluorescence techniques. As shown in Fig. 6C-D, the AHA@MnP NPs and AHA@MnP/QCT NPs significantly increased the secretion of ATP and HMGB1 in the cell culture medium. Also, CRT expression was significantly higher in lewis cells (possessing more fluorescent labeling) (Fig. 6E). The above results indicate that AHA@MnP/QCT NPs activate the cGAS-STING pathway by stimulating tumor cells to secrete ATP, HMGB1 and CRT, which elicits a strong anti-tumor immune response in the body.
Fig. 6
Mechanisms of immunogenic cell death in AHA@MnP/QCT NPs. (A) The expression of cGAS, p-STING and STING protein for dendritic cells treated with AHA@MnP or AHA@MnP/QCT NPs; (B) Quantitative analysis the protein intensity rate of dendritic cells. Data are presented as the mean ± SD (n = 3); (C) Intracellular ATP level for Lewis cells treated with AHA@MnP NPs or AHA@MnP/QCT NPs; (D) Intracellular HMGB1 level for Lewis cells treated with AHA@MnP NPs or AHA@MnP/QCT NPs. ****P < 0.001, ***P < 0.001, **P < 0.01, *P < 0.05 (E) Confocal images of CRT expression in Lewis cells pretreated with AHA@MnP NPs or AHA@MnP/QCT NPs (Scale bar: 50 μm)
In vivo antitumor assaysAHA@MnP/QCT NPs effectively induced ferroptosis and apoptosis in tumor cells in vitro and showed excellent targeting ability and accumulation at the tumor site. To evaluate their efficacy in vivo, we tested their ability to kill cancer cells in a Lewis tumor-bearing C57 mouse model. Mice were randomized into three groups once the tumor volume reached ~ 100 mm3 (n = 5 per group): (1) saline, (2) AHA@MnP NPs, and (3) AHA@MnP/QCT NPs. Group (1) received saline injections, while groups (2) and (3) received intravenous injections of their respective NPs on day 1 (Fig. 7A). Tumor size, body weight, and survival rates were recorded every 2 days to estimate the continuous therapeutic effectiveness. Compared with the Saline group, both NPs-treated groups showed significant tumor inhibition, with the AHA@MnP/QCT NPs demonstrating the most substantial anti-tumor effect (Fig. 7B-C). The tumor growth inhibition (TGI) rates for the saline, AHA@MnP NPs, and AHA@MnP/QCT NPs groups were − 857.39%, -430.86%, and − 310.90%, respectively. At the end of the treatment period, mice were executed, and tumors were harvested, showing a significant reduction in both tumor size and wight in the NPs-treated groups (Fig. 7D and G). Additionally, the AHA@MnP/QCT NPs group exhibited prolonged survival relative to the saline group (Fig. 7E). These results confirmed the superior anti-tumor efficacy of AHA@MnP/QCT NPs compared with saline.
To explore the mechanism of NPs’ anti-tumor effects, we also examined the distribution of immune cells in tumor sections and macrophage polarization. Fluorescent labeling and ImageJ quantification showed enhanced intertumoral abundance of dendritic cells, helper T cells (CD4 + T cells), and primary CTL (CD8 + T cells) in the AHA@MnP/QCT NPs and AHA@MnP NPs groups, along with a significant reduction in Treg cells due to Mn2+ overloading, compared with the saline group (Fig. 7F and H-K). These results suggest that AHA@MnP/QCT NPs can significantly elicit lymphocyte responses within tumors. Meanwhile, the results of polarization assays showed both NPs-treated tumor tissues had more M1-polarized macrophages (marked by CD80+) and fewer M2-polarized macrophages (marked by CD206+) relative to the saline group (Fig. 7F and L-M). The shift from M2 to M1 phenotypes suggests that AHA@MnP/QCT NPs activated anti-tumor immunity by inducing macrophage polarization [44]. One the other hand, significant effector T-cell infiltration was observed in the NPs-treated groups compared to the Saline group, likely due to the activation of the cGAS-STING signaling pathway by Mn2+ (Fig. 7F). Mn2+ overloading has been shown to increase the sensitivity of the cGAS pathway ini recognizing DNA, thereby promoting immune maturation [45]. The above results implied that NPs could inhibit tumors by activating immune responses, promoting tumor cell killing, and inhibiting tumor immune escape (Fig. 7N).
Fig. 7
In vivo antitumor effect of AHA@MnP/QCT NPs. (A) Scheme illustration for the in vivo evaluation of antitumor therapy; (B) The images for Lewis’s tumor bearing mice treated with AHA@MnP or AHA@MnP/QCT NPs; (C)Tumor growth curves of the mice treated with different samples. n = 5, ****P < 0.0001, ***P < 0.001; (D) Digital photos of excised tumors from the mice at day 12. (E) Survival curves analysis; (F) Immunofluorescence was used to examine CD11C+, CD4+, CD8+, Fopx3+, CD80 + and CD206 + in tumor sections. Scale bar = 50 μm; (G) Tumor weight analysis; (H-M) Fluorescence intensity of tumors calculated based on part (F) using ImageJ. The results are expressed as mean and SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; (N) Scheme illustration for AHA@MnP/QCT NPs involved in the immunotherapy process
留言 (0)