
記住我




The 344SQ parental (anti-PD1-sensitive) cell line (Sen) was a generous gift from Dr. Jonathan Kurie (MD Anderson). We used the Sen cell line to generate an anti-PD1-resistant cell line (Res) [30]. B16F10 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in complete medium (RPMI-1640 supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated fetal bovine serum) and incubated at 37 °C in 5% CO2. QPP7 glioblastoma cells were generated in the laboratory of Dr. Jian Hu and cultured in DMEM/F12 medium with B-27 supplement (Gibco), epidermal growth factor, and fibroblast growth factor (STEMCELL Technologies) [31]. Cell lines were validated by DDC Medical (http://ddcmedical.com; Fairfield, OH) by using short-tandem-repeat DNA fingerprinting.
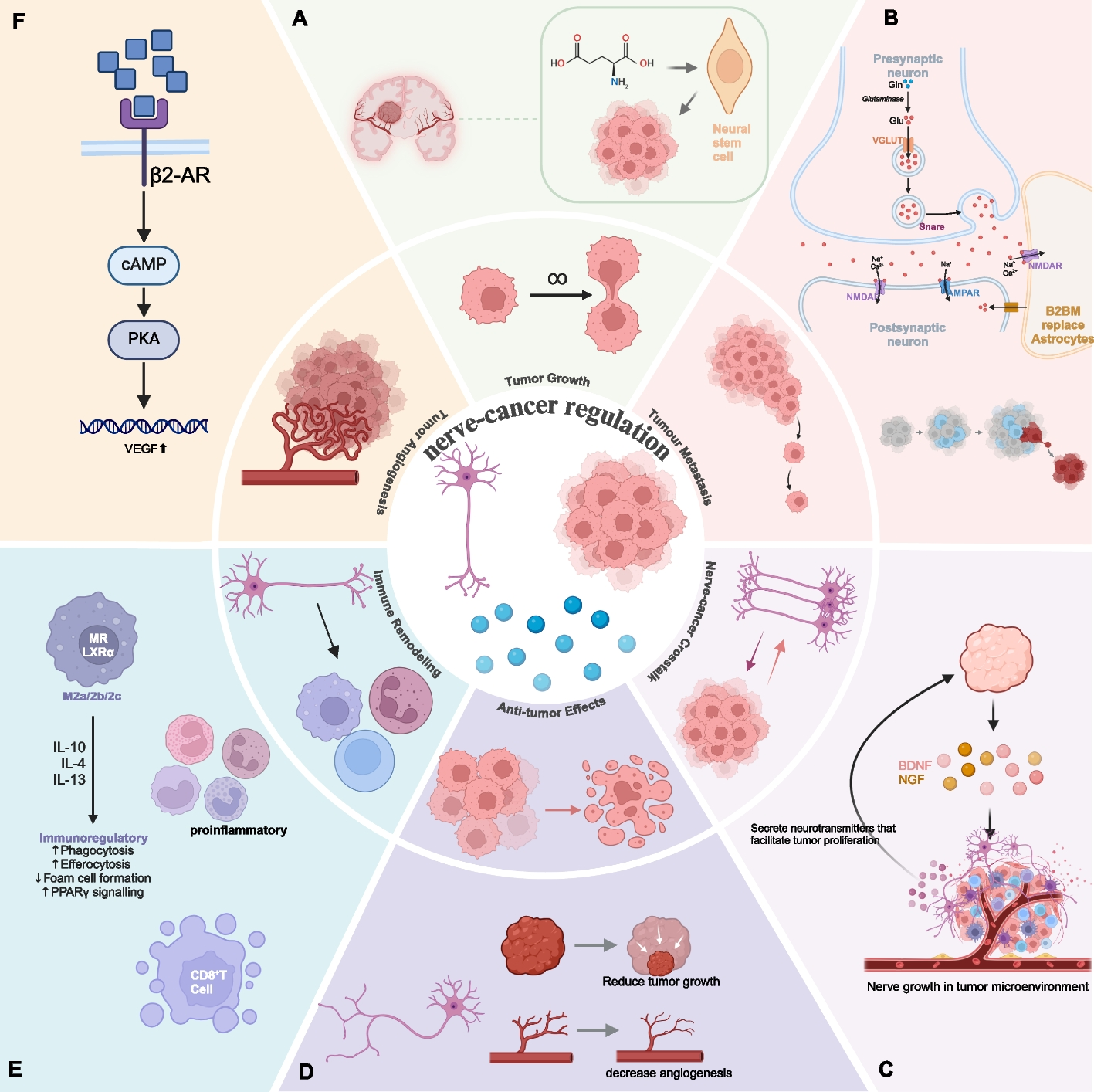
In vivo studiesAll mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Texas MD Anderson Cancer Center before their initiation; animal care was provided according to IACUC standards, and all mice were bred and maintained in our own specific pathogen-free mouse colony. Primary tumors were established by subcutaneous injection of Sen, Res, Res-ctrl, or Res-shFabp7 cells (0.5 × 106 in 100 μL of sterile phosphate-buffered saline [PBS]) into syngeneic 129 Sv/Ev mice (12–16 weeks old). B16F10-ctrl or B16F10-shFabp7 cells (0.5 × 106 in 100 μL of sterile PBS) were injected into syngeneic C57BL/6 mice (12–16 weeks old). Mice were then given intraperitoneal injections of anti-PD1 or control IgG antibodies (10 mg/kg; BioXcell) (n = 5 mice/group) starting on day 4 after tumor cell inoculation and continuing twice per week for a total of four doses. Rorafl mice were kindly provided by Dr. Cyrielle Billon and Thomas P. Burris and generated as described elsewhere [32]. To generate conditional knockout mice with Rora deletion specifically in CD8+ T cells, Rorafl mice were crossed with CD8-Cre transgenic mice (Jackson Laboratory, Stock No. 008766, C57BL/6-Tg(Cd8a-cre)1Itan/J). This cross resulted in offspring with the genotype Cd8 cre; Rorafl. Genomic DNA was extracted from tail biopsies of the offspring for genotyping. Polymerase chain reaction (PCR) was performed to confirm the presence of the floxed Rora alleles and the Cd8-Cre transgene. Primer sequences and PCR conditions are available upon request. The validation of the conditional knockout was performed by Transnetyx, Inc. (Cordova, TN). The Cd8 cre; Rorafl mice (12–16 weeks old) were subcutaneously injected with B16F10, B16F10-ctrl, or B16F10-shFabp7 cells (0.5 × 106 in 100 μL of sterile PBS), followed by intraperitoneal injections of anti-PD1 or control IgG antibodies (10 mg/kg; BioXcell) (n = 5 mice/group), starting on day 4 after tumor cell inoculation and continuing twice per week for a total of four doses. Tumors were measured with calipers three times per week and recorded as tumor volume (in mm3) = width2 × length/2. Tumor growth curves were compared with two-way analysis of variance. Tumor tissues were collected either at 24 h after the final anti-PD1 treatment (for lipidomics, global gene expression, qPCR, and immunohistochemical analyses) or at a week after the final anti-PD1 treatment (for isolating tumor-infiltrating lymphocytes [TILs]).
Unbiased lipidomicsLipid extraction, data acquisition by mass spectrometry, raw data processing, and data analysis were done by The Baylor College of Medicine Metabolomics Core and are all described elsewhere [33]. Briefly, samples were extracted with water / methanol / dichloromethane (2:2:2 v/v), with mouse liver samples as a quality control to monitor the extraction efficiency and instrument performance. Lipids were separated by reverse-phase chromatography (Acquity HSS UPLC T3 column [1.8 μm particle 50 × 2.1 mm], Waters, Milford, MA) on a Shimadzu CTO-20A Nexera X2 UHPLC system, with LC mobile phase solvent A acetonitrile/water (40:60, v/v) with 10 mM ammonium acetate and solvent B acetonitrile / water / isopropanol (10:5:85 v/v) with 10 mM ammonium acetate. The flow rate for these experiments was 0.4 mL/min. Mass spectrometry data were acquired by Data Independent Acquisition (DDA) methods in both positive and negative ionization modes by using a TripleTOF 5600. Identified peaks and retention time were carefully reviewed with MultiQuant software (ver. 1.1.0.26, AB Sciex, Concord, Canada). The relative peak area was log2 transformed followed by internal standard normalization for each method. Analysis of differentially expression between groups was done with the Benjamini–Hochberg method for false discovery rate (FDR) of < 0.25 correction to account for multiple comparisons.
Lipid (Oil Red O) stainingPD1-sensitive (Sen) and PD1-resistant (Res) cells were counted and seeded into 6-well plates at a density of 300,000 cells per well. After 20 h, neutral lipids within lipid droplets were quantified by using an Oil Red O Staining Kit (Catalog #MAK194, Sigma) specifically designed for cultured cells according to the manufacturer's instructions. For in vivo experiments, tumors were harvested from mice that had been subcutaneously injected with either Sen (n = 3) or Res (n = 3) cells at a concentration of 500,000 cells per mouse. Mice were then treated twice weekly for 2 weeks with either IgG or PD1 antibody after tumor implantation. Tumors were snap-frozen in optimal cutting temperature compound and preserved at −80 °C immediately after collection. Tissue sections (thickness 5 µm) were stained for lipid content by using an Oil Red O Stain Kit (Catalog #ab140678, Abcam) per the manufacturer's guidelines. Slides were examined with a Leica DMI6000B microscope (Leica, Buffalo Grove, IL), and images were captured with a charge-coupled device camera.
Targeted lipidomics mass spectrometry for eicosanoids/oxylipinsTwelve tumors were obtained from mice that had been injected subcutaneously with anti-PD1-sensitive (Sen) (n = 3) or PD1-resistant (Res) (n = 3) cells (500,000 cells/mouse), followed by twice-weekly treatment with either IgG or PD1 antibody after implantation for 2 weeks. Tumors were collected, snap-frozen immediately, and stored at –80 °C until analysis processing and analysis by Creative Proteomics, Shirley, NY). Sample extraction and cleanup was done according to a protocol outlined by Watrous [34]. Briefly, each tumor was individually weighed, and 10 ng of d8-arachidonic acid or d4-resolvin e1 were added to each. Samples were then processed by adding 250 μL of –20 °C chilled 75% ethanol and homogenized in a ThermoFisher bead mill for 2 min. The homogenates were transferred to 2.0 mL centrifuge tubes, to which an additional 750 μL of –20 °C chilled 75% ethanol was added. Samples were vortexed for 30 min, incubated at –20 °C for 1 h to precipitate proteins, and centrifuged at 15,000 × g for 20 min. The supernatants were collected, and the protein pellets were re-extracted with the supernatants pooled. The pooled supernatants were then diluted to 10 mL with HPLC water, applied to preconditioned solid-phase extraction (SPE) columns, washed, and eluted as per the established protocol. After the SPE process, samples were dried under vacuum, reconstituted in acetonitrile, and stored at –80 °C until analysis. For LC–MS analysis, we used a Shimadzu Prominence HPLC coupled to a Thermo LTQ-Orbitrap Velos mass spectrometer. The LC system included a specific HPLC column and gradient conditions adapted from Watrous [34], with the mass spectrometer operated in negative ion mode under specific settings. Peaks were identified and quantified with MAVEN software, with concentrations normalized to the weight of the individual tumor and compared against reference standards for confirmation. Data were analyzed with Thermo Scientific LipidSearch software (version 5.0) and R scripts written by the Metabolomics Core Facility at MD Anderson Cancer Center.
Long-chain fatty acid oxidation stress testOxygen consumption rates (OCR) of the mitochondria in PD1-sensitive (Sen) and PD1-resistant (Res) cells were measured by a Seahorse XFe24 Analyzer (Agilent Technologies) and an XF Long Chain Fatty Acid Oxidation Stress test kit (catalog #103,672–100, Agilent Technologies). Briefly, cells were seeded and cultured in XFe24 cell culture plates; for the experiments, the medium was replaced with pre-warmed assay medium (Seahorse base medium supplemented with 1 mM pyruvate, 10 mM glucose, 2 mM glutamine, and 4 uM Etomoxir, pH = 7.4) and the plate were incubated at 37 °C in a non-CO2 incubator for 1 h. Next, plates were transferred to the Seahorse XFe24 to record the OCR of the cells at different times. The OCR readings were normalized to the cell numbers in each well. Mitochondrial respiration, including basal respiration, proton leak, and maximal respiration were calculated by the Seahorse Wave software.
Global mRNA expression profilingPD1-sensitive (Sen) or PD1-resistant (Res) cells were inoculated into the right flank of 129 Sv/ev mice (female, 12–14 weeks old, 5 mice per group). Anti-PD1 or control IgG antibodies (10 mg/kg) were given on days 4 and 7 after tumor inoculation. On day 11, tumor tissues were collected, immediately frozen in liquid nitrogen, and homogenized with grinders in the presence of Trizol (Qiagen). Total RNA was isolated by phenol-based extraction and resolved in water treated with diethylpyrocarbonate. The purity and concentration of total RNAs were determined with a Nanodrop 1000 spectrophotometer. The total RNA samples were further analyzed with Agilent’s 2100 Bioanalyzer to assess sample quality and integrity. RNAs from three independent biological replicates per group were used for GeneChip Mouse genome 430 2.0 arrays (Affymetrix). Sample labeling and processing, GeneChip hybridization, and scanning were done according to Affymetrix protocols (3’ IVT plus reagent labeling kit). Each array was washed and stained with streptavidin–phycoerythrin (Invitrogen) and amplified with biotinylated anti-streptavidin antibody (Vector Laboratories) on a GeneChip Fluidics Station 450 (Affymetrix). Arrays were scanned with the GeneArray G7 scanner (Affymetrix) to obtain image and signal intensities. After scanning, the images were processed by using Affymetrix Expression Console Software Version 1.0 to generate gene expression intensity values. Differentially expressed genes were identified by using parametric tests with log2-transformed gene expression values. P values obtained were adjusted using the false discovery rate (Benjamini–Hochberg). Heatmaps were created by using the heatmap.2 function in the gplots R package. To identify pathways or functions that were overrepresented among the genes at the top or bottom of the ranked gene list, we used the pre-ranked gene set enrichment analysis method.
Quantitative polymerase chain reactionTotal RNA was isolated from cells and tumors with Triazol (Life Technologies) according to the manufacturer’s protocol. For studies of Fabp7, Pparg, Hif1a, Rora, Bmal1 (Arntl), Clock, Nrip1, Bcl2l11, CD45 and Rps18 expression, mRNA was retrotranscribed with an iScript gDNA Clear cDNA Synthesis Kit (BioRad, catalog# 1,708,890) or High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific, catalog# 4,368,814) and analyzed by quantitative PCR by using SYBR Green (Life Technologies) with specific primers (Supplementary Table S1) according to the manufacturer’s protocol. The comparative Ct method was used to calculate the relative abundance of mRNAs compared with Rps18 or CD45 expression.
Immunohistochemical analysisFormalin-fixed mouse tissues were processed at the MD Anderson Research Histology Core Laboratory. Briefly, tissues were submitted on an automatic tissue processor, embedded in paraffin (Peloris, Leica), and cut into 4-μm sections. Immunohistochemical staining was done with an automated staining system (Leica Bond Max, Leica Microsystems, Vista, CA, USA) as follows. Briefly, slides were deparaffinized and hydrated, and antigen was retrieved by incubating in citrate buffer (pH 6.0) for 1 h with Fabp7 (ThermoFisher, Catalog #PA5-23,469) and 4-hydroxynonenal (4-HNE) antibody (Abcam, Catalog #ab48506) according to the manufacturer’s protocol. Slides were examined with a Leica DMI6000B microscope (Leica, Buffalo Grove, IL), and images were captured with a charge-coupled device camera, imported into the Advanced Spot Image analysis software package, and quantified with Fiji software (http://fiji.sc).
Stable Fabp7-knockdown and control cellsTo establish stable Fabp7-knockdown cells, GIPZ Non-silencing Lentiviral shRNA Control (Catalog #RHS4348, Dharmacon) and specific mouse shRNA targeting Fabp7 (pGIPZ Clone ID V2LMM_50996, Dharmacon) viral supernatants were purchased from the shRNA and ORFeome Core at MD Anderson Cancer Center. Res cells were infected and incubated with the viral particles supplemented with polybrene (8 µg/mL, Sigma) overnight at 37 °C. Puromycin (1 µg/mL) was used to select and maintain Fabp7-knockdown in Res, B16F10, and QPP7 cells. Stable repression was verified by quantitative PCR and western blotting.
PPARγ transcription factor assayPPARγ activation was evaluated by using a PARγ Transcription Factor Assay kit (catalog #ab133101) according to the manufacturer’s protocol. To initiate the assay, a specific double-stranded DNA sequence containing the peroxisome proliferator response element (PPRE) was immobilized onto the bottom wells of a 96-well plate. Subsequently, nuclear extracts from Sen, Res, ctrl, or shFabp7 cells containing PPARs were introduced into the wells; PPARγ specifically bound to the immobilized PPRE was detected by adding a specific primary antibody targeted to PPARγ. A secondary antibody conjugated to horseradish peroxidase was then applied to enable a sensitive colorimetric readout at 450 nm with a BioTek Sinergy plate reader (Agilent Technologies).
Enzyme-linked immunosorbent assayEnzyme-linked immunosorbent assay (ELISA) was used to measure Fabp7 levels in the culture supernatants. Supernatants were freshly collected from four different cell types: Sen, Res, Res-ctrl, and Res-shFabp7, and were immediately processed for analysis. The Fabp7 concentration was quantified by using an ELISA kit according to the manufacturer's instructions (LSBio, Catalog # LS-F11416). The absorbance readings necessary for determining Fabp7 levels were obtained with a BioTek Sinergy plate reader (Agilent Technologies).
Chromatin immunoprecipitation sequencing (ChIP-Seq)Samples from Sen, Res, Res-ctrl and Res-shFabp7 cells were processed for H3K27ac and H3K9ac ChIP and ChIP-Seq libraries were prepared at the MD Anderson Epigenomics Profiling Core Facility. The ChIP-Seq libraries, along with the corresponding input libraries, were sequenced by using a 50-base single-read protocol on an Illumina NovaSeq 6000 instrument at the MD Anderson Advanced Technology Genomics Core. In total, 24 ChIP-Seq libraries (three biological replicates per histone mark and condition), as well as the corresponding input libraries, were sequenced, generating 53–158 million reads per sample. For mapping, the reads were mapped to the mouse genome (mm10) by using Bowtie (version 1.1.2) with the following parameters: "-v 2 -m 1 –best –strata". To avoid PCR bias, only one copy of multiple reads mapped to the same genomic position was retained for further analysis. For peak calling, peaks were identified on each ChIP-Seq sample by using MACS (version 1.4.2) with comparison against the corresponding input sample. A window size of 500 bp was used, and a p value cutoff of 1e-5 was applied. Peaks that overlapped with ENCODE blacklisted regions were removed. For each differential comparison, peaks from all the involved samples were merged, and the number of reads within these merged peaks was counted for each sample. Merged peaks with less than 10 reads in all samples were removed. The resulting count table was used to identify differential peaks by using the R/Bioconductor package edgeR. The numbers of reads within the common peaks of all samples were used as the library sizes in edgeR. Peaks with a false discovery rate (FDR) of ≤ 0.05 and a fold change of ≥ 1.5 or 2 were identified as differential peaks and presented in heatmap plots. For signal tracking, each read was extended by 150 bp to its 3’ end. The count of reads covering each genomic position was multiplied by 1 × 107 divided by the library size used in edgeR. These values were then averaged over a 10 bp resolution. The resulting averaged values were displayed by using the Integrative Genomics Viewer. To generate heatmaps, the region spanning 10 kb upstream to 10 kb downstream from the center of each differential peak was divided into 250 bp bins. The number of reads within each bin was multiplied by 1 × 10–6, divided by the library size used in edgeR, and then averaged across replicate samples. The resulting value tables were then visualized in heatmap by using the R function heatmap.2. Motif enrichment was anayzed with The MEME Suite 5.5.5 [35], and Giggle score was obtained with the genomic search engine GIGGLE (https://github.com/ryanlayer/giggle) [36].
Untargeted lipidomic analysis by LC–MS/MSSamples were sent to Metabolomics Core Facility at the MD Anderson for processing and analysis. Around 10 million cells were harvested for each run as instructed by the core facility. Briefly, upon harvest, cells were washed with ice-cold 0.85% ammonium bicarbonate in deionized water. Cells were then scraped down and pelleted at 400 × g for 3 min. The supernatant was removed, and the cells were snap-frozen in liquid nitrogen and kept in a –80ºC freezer before being sent to the Metabolomics Core Facility. To determine the relative abundance of lipid in different prostate cancer cells, extracts were prepared and analyzed by high-resolution mass spectrometry-based lipidomics at the Metabolomics Core Facility. Briefly, to each cell sample, 200 µL of extraction solution containing 2% Avanti SPLASH® LIPIDOMIX® Mass Spec Standard, 1% 10 mM butylated hydroxytoluene in ethanol was added and the tubes were vortexed for 10 min. The tubes were placed in ice for 10 min and centrifuged at 13,300 rpm for 10 min at 4ºC. The supernatant was transferred to a glass autosampler vial, and the injection volume was 10 µL. Mobile phase A (MPA) was 40:60 acetonitrile: water with 0.1% formic acid and 10 mM ammonium formate. Mobile phase B (MPB) was 90:9:1 isopropanol:acetonitrile: water with 0.1% formic acid and 10 mM ammonium formate. The chromatographic method included a Thermo Fisher Scientific Accucore C30 column (2.6 µm, 150 × 2.1 mm) maintained at 40 °C, autosampler tray chilling at 8 °C, a mobile phase flow rate of 0.200 mL/min, and a gradient elution program as follows: 0–3 min, 30% MPB; 3–13 min, 30–43% MPB; 13.1–33 min, 50–70% MPB; 48–55 min, 99% MPB; 55.1–60 min, 30% MPB. A Thermo Fisher Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer with heated electrospray ionization source was operated in data-dependent acquisition mode, in both positive and negative ionization modes, with scan ranges of 150–827 and 825–1500 m/z. An Orbitrap resolution of 120,000 (FWHM) was used for MS1 acquisition, and a spray voltage of 3,600 and –2900 V were used for positive and negative ionization modes. Vaporizer and ion transfer tube temperatures were set at 275 and 300 °C, respectively. The sheath, auxiliary and sweep gas pressures were 35, 10, and 0 (arbitrary units), respectively. For MS2 and MS3 fragmentation, a hybridized HCD/CID approach was used. Each sample was analyzed by using four injections, making use of the two aforementioned scan ranges, in both ionization modes. Data were analyzed with Thermo Scientific LipidSearch software (version 5.0) and R scripts written in-house.
Reactive oxygen species assayResistant control (Res-ctrl) and Resistant shFabp7 (Res-shFabp7) cells were cultured under standard conditions. For the assay, 10,000 cells from each line were counted and plated into individual wells of a 96-well plate. Cellular oxidative stress was analyzed with CellROX Deep Red Reagent (Thermo Fisher Scientific, Catalog #C10422), a cell-permeant dye that is non-fluorescent under reduced conditions and exhibits fluorescence when oxidized by ROS. The dye has absorption and emission maxima at approximately 644 / 665 nm. The Incucyte SX1 Live-Cell Analysis System (Sartorius) was used for monitoring the cells over a period of 40 h. Image acquisition was automated and set at regular intervals during this period. Images were analyzed with Incucyte software, which provided quantitative data on the levels of oxidative stress in the cells.
Ex vivo co-culture of CD8+ T cells with cancer cellsViable cells were quantified with a hemocytometer with a 0.4% Trypan blue solution, and subsequently diluted to a density of 300,000 cells per well in 6-well plates. Res-ctrl or Res-shFabp7 cells were seeded in the upper inserts (24.5-mm Transwell with 0.4-µm pore polycarbonate membrane inserts, Fisher Scientific), whereas CD8+ T cells were placed in the lower chamber of the transwell system. CD8+ T cells were isolated from splenocytes before seeding by using the Dynabeads Untouched Mouse CD8 Cells Kit (Thermo Fisher Scientific–Life Technologies, Catalog #11417D). The cells were then activated with Ultra-LEAF Purified anti-mouse CD3ε Antibody (5 μg/mL) (Biolegend, Catalog # 100,301) and LEAF purified anti-mouse CD28 antibody (1 μg/mL) (Biolegend, Catalog # 102,101). Cells were cultured in complete medium (RPMI-1640 supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated fetal bovine serum), and incubated at 37 °C in a 5% CO2 for 24 or 48 h. Subsequently, RNA was isolated from the CD8+ T cells for gene expression analysis by quantitative PCR or Western blotting.
Assay for transposase-accessible chromatin with sequencing (ATAC-Seq)ATAC-Seq libraries were prepared from T cells cocultured with Res-ctrl or Res-shFabp7 for 48 h at the MD Anderson Epigenomics Profiling Core Facility. The ATAC-Seq libraries were sequenced by using a 2 × 76 base paired-end protocol on an Illumina NovaSeq 6000 instrument at the MD Anderson Advanced Technology Genomics Core. In total, six ATAC-Seq libraries (three biological replicates per condition) were sequenced, generating 35–75 million pairs of reads per sample. Each pair of reads represents a DNA fragment from the library. For mapping, adapter sequences were removed from the 3' ends of reads by using Trim Galore! (version 0.6.5) and cutadapt (version 2.8). The reads were then mapped to the mouse genome (mm10) with Bowtie (version 1.1.2) with the following parameters: "–allow-contain –maxins 2000 -v 2 -m 1 –best –strata". To avoid PCR bias, only one copy of multiple fragments mapped to the same genomic position was retained for further analysis. After the removal of fragments from chrM, for each fragment, the 5’ end was offset by + 4 bp and the 3’ end was offset by −5 bp to adjust both ends to represent the center of a transposon binding event. For peak calling, peaks were identified for each sample by using MACS2 (version 2.2.7.1) without any control. Each binding event (i.e., the 5’ or 3’ end of a fragment) was smoothed by 73 bp (i.e., extended 36 bp upstream and 36 bp downstream from the event center). MACS2 was configured to call peaks from the pile-up of smoothed binding events, and a q-value cutoff of 0.05 was applied. The peaks that overlapped with ENCODE blacklisted regions were removed. For differential peak analysis, peaks from all the samples were merged, and the number of transposon binding events within these merged peaks was counted for each sample. Merged peaks with less than 10 binding events in all samples were removed. The resulting count table was used to identify differential peaks with the R/Bioconductor package edgeR. The numbers of binding events within the common peaks of all samples were used as the library sizes in edgeR. Peaks with a false discovery rate (FDR) of ≤ 0.05 and a fold change of ≥ 2 were identified as differential peaks and presented in heatmap plots. For signal tracking, each transposon binding event was smoothed to a length of 73 bp, spanning from −36 bp to + 36 bp around the center. The count of binding events covering each genomic position was multiplied by 1 × 107 divided by the library size used in edgeR. These values were then averaged over a 10 bp resolution. The resulting averaged values were displayed by using the Integrative Genomics Viewer. To generate heatmaps, the region spanning 10 kb upstream to 10 kb downstream from the center of each differential peak was divided into 250 bp bins. The number of transposon binding events within each bin was multiplied by 1 × 10.6, divided by the library size used in edgeR, and then averaged across replicate samples. Subsequently, the resulting value tables were visualized in heatmap by using the R function heatmap.2. Motif enrichment was analyzed with The MEME Suite 5.5.5 [35], and the Giggle score was obtained with the genomic search engine GIGGLE (https://github.com/ryanlayer/giggle) [36].
RNA sequencingT cells were co-cultured with cancer cells as previously described. After 48 h, T cells were collected, and RNA was isolated for RNA-seq analysis by using Trizol according to the manufacturer’s protocol. RNAseq sample quality control was done with FastQC. Sequencing reads were aligned to Genome Reference Consortium Human Build 38 (GRCh38.p13) (Genome Reference Consortium Mouse Build 39 [GRCm39]) by using STAR. The expression abundance and variations of mRNA were calculated as expected counts and transcripts per million (TPM) by using RSEM software. Differentially expressed genes were identified with parametric tests and log2-transformed TPM values. P values obtained from the tests were adjusted by using the false discovery rate (Benjamini–Hochberg). To identify pathways or functions that were overrepresented among the genes at the top or bottom of the ranked gene list, we used the pre-ranked Gene Set Enrichment Analysis method and Gene Ontology (https://geneontology.org/).
Protein extraction and western blot analysisTotal protein was extracted by using NP40 lysis buffer (0.5% NP40, 250 mmol/L NaCl, 50 mmol/L HEPES, 5 mmol/L ethylenediaminetetraacetic acid, and 0.5 mmol/L egtazic acid) supplemented with protease inhibitors cocktails (Sigma-Aldrich). Lysates were centrifuged at 10,000 × g for 10 min, and the supernatant was collected for experiments. Protein lysates (40 μg) were resolved on denaturing gels with 4%–20% sodium dodecyl sulfate–polyacrylamide and transferred to nitrocellulose membranes (BioRad Laboratories, Hercules, CA). Membranes were probed with primary antibodies directed against vinculin (Millipore Sigma, Catalog #05–386), p53 (Millipore Sigma, Catalog #OP43) and FABP7 (ThermoFisher, Catalog# PA5-31,864), (dilution 1:500), and a secondary antibody conjugated with horseradish peroxidase (dilution 1:2000) (Amersham GE Healthcare). The secondary antibody was visualized by using a chemiluminescent reagent (Pierce ECL kit, Thermo Fisher Scientific, Waltham, MA, USA).
Isolation of tumor-infiltrating lymphocytesTumor-infiltrating leukocytes (TILs) were isolated from freshly extracted primary tumor tissues (obtained from three mice per group) by using a Tumor Dissociation Kit (Miltenyi, Catalog # 130–096–730) in conjunction with the gentleMACS Octo Dissociator with Heaters (Miltenyi, Catalog #130–096–427), according to the manufacturer's protocol. After dissociation, TILs were enriched by using a Dynabeads Untouched Mouse T Cells Kit (Thermo Fisher Scientific–Life Technologies, Catalog #11413D). The enriched TILs were subsequently used for both quantitative PCR and flow cytometry analysis.
Co-immunoprecipitation assayCell lysates from Res cells treated with dimethylsulfoxide or DHA (30 μg/mL) (Cayman Chemicals, catalog #90,310) were processed for immunoprecipitation analysis by Creative Proteomics (Shirley, NY). Briefly, cell lysates were first precleared by adding 1 µg of anti-Fabp7 antibody (Abcam, Catalog# ab279649) and 20 µL of Protein A/G PLUS-Agarose. This mixture was incubated at 4 °C for 30 min. After the incubation, the lysate-beads mixture was centrifuged at 3,000 rpm for 5 min at 4 °C, and the supernatant was transferred to a fresh 1.5 mL conical tube and kept on ice. Next, 20 µL of anti-Fabp7 antibody was added to 500 µg of the cell lysate in a 1.5 mL microcentrifuge tube, which was incubated for 1 h at 4 °C. Subsequently, 20 µL of Protein A/G PLUS-Agarose was added to the mixture, which was incubated overnight at 4 °C on a rotating device. The immunoprecipitated samples were collected by centrifugation at 3,000 rpm for 5 min at 4 °C, after which the supernatant was carefully aspirated and discarded. The beads were washed four times with PBS, with centrifugation repeated after each wash. After the final wash, the supernatant was aspirated and discarded, and the beads were resuspended in 40 µL of 1X electrophoresis sample buffer. Finally, the samples were boiled for 15 min and prepared for Western blotting by using Rora antibody (Abcam, Catalog #ab256799).
Flow cytometryTo block Fc receptor-mediating binding, cells were pre-incubated with anti-CD16/CD32 antibody prior to staining (1:200). For flow cytometry purposes, fluorochrome-conjugated anti-CD3 (Cat #100353), anti-CD45 (Cat #103126), and anti-CD8 (Cat #100734) antibodies were purchased from BioLegend. Apoptosis in CD8+ T cells was analyzed with an APC Annexin V Apoptosis Detection Kit with PI (Biolegend, Catalog # 640932). Samples were stained according to the manufacturer’s protocol and analyzed with an LSR II flow cytometer and FlowJo software (version 10.10).
Statistical analysisStatistical analyses were done with R (version 4.0.1). An unpaired t test was used to compare the mean of two different groups when the distribution of the population was normal. One-way analysis of variance was used to compare the means of three or more groups under the assumption of normal distribution, followed by Tukey's HSD (honestly significant difference) test. The nonparametric Mann–Whitney U test and Kruskal–Wallis H test were used to compare the mean ranks between two groups (U test) or three groups (H test). Student’s t tests (two-tailed) were used to compare differences between individual groups, with error bars representing the standard deviation. P values were adjusted for multiple hypothesis testing by using Benjamini–Hochberg method when needed. Statistical significance was indicated by p < 0.05.
留言 (0)