Recombinant antigen expression
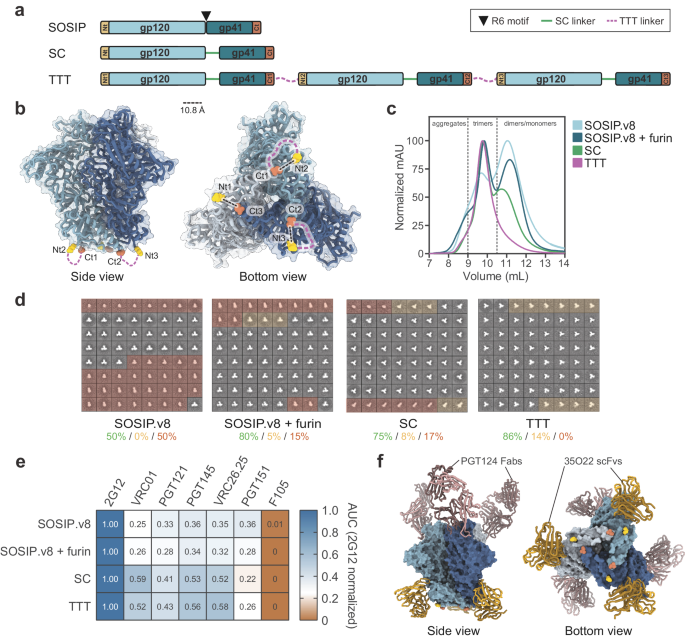
Production of soluble, recombinant E2 (sE2) antigen was based on the Con1 (genotype 1b) isolate sequence (amino acids 384–661)35,49, which was truncated in the C-terminal transmembrane stem region. The gene sequence was modified to contain an N-terminal BIIP sequence as well as a C-terminal 6x poly-histidine (HIS) tag followed by a SpyTag (Fig. 1A). The final gene had flanking restriction sites (EcoRI and NotI) added to the N- and C-terminus and was codon optimized for expression in Drosophila Schneider 2 (S2) insect cells. The gene was synthesized by GeneArt (Thermo Scientific) and subcloned into the pExpreS2-1 (zeocin resistance) vector (ExpreS2ion Biotechnologies, Horsholm, Denmark) using EcoRI and NotI restriction sites.
The S2 cell culture supernatant was concentrated five times and buffer exchanged into 20 mM Tris, 0.5 M NaCl, pH 7.9 using a tangential flow filtration system (Quixstand Benchtop system, MWCO 10 kDa). Concentrated and buffer exchanged supernatant was loaded onto 5 mL HisTrapTM HP columns for ion metal affinity chromatography (IMAC) and purified using the above buffer with 60 mM Imidazole and increasing to 0.5 M Imidazole in the elution buffer for step elution of the bound protein. The elution peak was further purified by size exclusion chromatography (SEC) on a 200 pg Sephadex column. Post purification SEC fractions of peak 1 and 2 (Fig. 1C) were used for subsequent immunization studies as monomeric and oligomeric sE2 antigens, respectively.
AP205 cVLP production
The SpyCatcher-AP205 cVLP is assembled from fusion proteins comprising the major capsid protein of the Acinetobactor phage AP205 (Gene ID: 956335) and SpyCatcher (Genbank: AFD50637, aa 24-139), respectively. Specifically, the SpyCatcher-AP205 sequence was constructed by fusing the SpyCatcher sequence to N-terminus of the AP205 coat protein, using a flexible linker (Gly-Gly-Ser-Gly-Ser), and subsequently adding flanking NcoI and NotI restriction sites at the N- and C-termini, respectively. The gene sequence was finally codon-optimized for recombinant expression in E. coli and synthesized by (GeneArt® Life Technologies, Germany). The synthetic gene was cloned into a pET-15b vector and transformed into competent One Shot® BL21 Star™ (DE3) cells (Thermo Scientific). Recombinant protein expression was done in 3 L shake flasks containing 400 mL 2xYT media (100 µg/mL ampicillin). The bacterial culture was incubated at 37 °C for three hours (OD600 = 0.6) at which point cells were induced with 1 mM IPTG. The induced culture was then incubated for additional 16 h at 20 °C. Bacterial cells were harvested by centrifugation (10,000 × g) and the pellet was resuspended in 1× PBS (pH = 7.2) and lysed by sonication at 80% power with 5 pulsations for 2 × 5 min on ice. The cleared bacterial lysate was finally purified by ultracentrifugation (UC) using an Optiprep™ (Sigma) step (23, 29, and 35%) gradient. Specifically, 1.2 ml lysate was loaded on top of the density step-gradient and spun at 307.900 RCF (SW60Ti rotor, Beckmann Coulter) for 3.30 h at 16 °C. The UC sample was then divided into smaller fractions (~200 µl), which were analyzed for protein content by SDS-PAGE. Fractions containing SpyCatcher AP205 cVLPs were finally pooled and dialyzed against 20 mM Tris, 0.5 M NaCl buffer using a 1000 kDa MWCO SpectraPor Membrane.
Vaccine production
Purified, SpyTagged monomeric or oligomeric sE2 protein was mixed with SpyCatcher-AP205 cVLP at molar ratio 2:1 (antigen per cVLP binding site) and incubated for 36 h at 4 °C to generate sE2-cVLPs (Fig. 1B). Analysis of the vaccine mixture was done by SDS-PAGE. Briefly, 10 µL of the vaccine sample was mixed with 2 µL SDS loading dye (with or without Dithiothreitol). After heating the sample at 95 °C for 5 min, it was added (with a molecular size marker) to a NuPAGE Bis-Tris gel and run for 1 h at 170 V. The SDS-PAGE gel was finally stained with coomassie blue. SDS-PAGE densitometric analysis (ImageQuantTL) was used to determine the coupling efficiency of the SpyTag-SpyCatcher interaction by dividing the intensity of the cVLP band in the coupling lane with the intensity of the cVLP band in an input lane containing the equivalent amount of cVLP used in the coupling reaction and multiplying this value with 100. Excess unbound antigen was removed by repeating the ultracentrifugation step. sE2-cVLP containing fractions were pooled and dialyzed against 20 mM Tris, 0.5 M NaCl, pH7.9 as above and bacterial endotoxins were removed from the non-displayed and cVLP-displayed vaccines by a phase separation method using Triton X-114 detergent. Specifically, Triton X-114 was added in a 1:100 volume ratio to the protein sample, which had been chilled on ice. The sample was then mixed by vortexing and incubated on ice for 5 min. Subsequently, the sample was incubated at 37 °C for 5 min and centrifuged for 1 min at 16.000 × g at 37 °C. After separating the supernatant from the pellet, the above procedure was repeated using only the supernatant. Triton X-144 was finally removed by dialysis. All gel electrophoresis lanes shown in Coomassie stains of SDS-PAGE were run at the same time.
Transmission electron microscopy (TEM)
For TEM imaging, monomeric and oligomeric sE2-cVLP vaccines samples were diluted to 0.2 mg/mL in PBS and adsorbed to 200-mesh-carbon-coated grids, which were stained with 2% phosphotungstic acid (pH = 7.0) for 1 min. The negatively stained sample was finally analyzed in a CM100 BioTWIN electron microscope (Phillips) at an accelerating voltage of 80 kV. Pictures were taken using an Olympus Veleta camera.
Dynamic Light Scattering (DLS)
DLS was used to investigate the distribution of cVLP particle sizes. Samples were spun for 2.5 min at 16.000 RCF before analysis. Twenty repeated measurements were acquired for each sample (at 658 nm, 25 °C, WYATT Technology, DynaPro NanoStar). The diameter was calculated from the hydrodynamic radius of the particles along with percent polydispersity (%Pd).
Animal immunizations
Mice were immunized with monomeric or oligomeric HCV sE2, alone or displayed on AP205 cVLP, to evaluate the immunogenicity and capacity for inducing NAbs of the different vaccines. Each immunization group contained six female BALB/c mice (8 weeks old) that were immunized intramuscularly in the thigh at weeks 1, 3, and 6. Mouse blood samples were taken at weeks 0 (pre-bleed), 6, and 9 (Fig. 2A). Per immunization, each mouse received 1.5 µg of either non-displayed or cVLP-displayed sE2 antigen formulated 1:1 (vol/vol) in Addavax (Invivogen).
HCV sE2 specific serum IgG levels
To determine the level of HCV sE2-specific antibody titers raised following immunization with both monomeric and oligomeric variants of the sE2 antigen displayed on cVLP or non-displayed, an enzyme-linked immunosorbent assay (ELISA) was used. Specifically, 96-well microtiter plates (Nunc MaxiSorp) were coated over night at 4 °C with monomeric or oligomeric sE2 with C-terminal HIS-Tag (0.1 µg per well) produced in insect cells. PBS buffer +0.5% skim milk powder (w/v) was added for blocking of the plates. PBS was used for all washing steps. Secondary goat anti-mouse IgG Horseradish peroxidase (Novex) (1:2000 dilution) was used in as secondary antibody and 3,3′,5,5′-Tetramethylbenzidine (TMB) as substrate for developing the plates. The colorimetric reaction was terminated with 0.2 M H2SO4 and the signal was measured at OD 450 nm. IgG titers were normalized across plates using a positive control and antibody titers were calculated by determining OD50 (after modeling the serum dilution curves in Graphpad Prism (9.2.0) software using four parameter non-linear regression curve fitting).
Competition ELISA against AR1B, AR2A and AR3A
96-well microtiter plates (Nunc MaxiSorp) were coated with monomeric sE2 at 0.4 µg/ml ON at 4 °C. The following day, the plates were washed with PBS 0.1%Tween20 and blocked using PBS buffer with 1% skim milk powder (w/v) and 5% BSA (BSK). Three-fold dilution series of serum were made in PBS 0.1%Tween20 starting at 1:200 and incubated with the plates ON at 4 °C. The following day, human monoclonal antibodies AR1B, AR2A, and AR3A were used at 50% binding concentrations and incubated in BSK for 30 min at room temperature. After washing in PBS 0.1%Tween20 sheep anti-human secondary antibody coupled to HRP (NA933V) was incubated at 1:1000 dilution in BSK for 1 h at room temperature. 3,3′,5,5′-Tetramethylbenzidine (TMB) was used as substrate for developing the plates and the colorimetric reaction was terminated with 2 M H3PO4 and the signal was measured at OD 450 nm.
Antigenic sE2-cVLP characterization by ELISA
96-well microtiter plates (Nunc MaxiSorp) were coated with cVLPs displaying monomeric or oligomeric sE2 (0.1 µg per well). Dilution series of HCV E2-specific human monoclonal antibodies AR1B, AR2A, or AR3A24 were subsequently tested for binding to the immobilized E2-cVLP complexes to probe epitope accessibility. PBS buffer +0.5% skim milk powder (w/v) was used for blocking the plates and for serum dilutions. PBS was used for all washing steps. Secondary goat anti-human IgG Horseradish peroxidase (Agilent Technologies) was used as secondary antibody (1:5000 dilution) and TMB as substrate for developing the plates. The colorimetric reaction was terminated with 0.2 M H2SO4 and the signal was measured at OD 450 nm.
Mouse IgG purification and quantification
For the neutralization experiments, mouse IgG was purified from serum taken at 3 weeks after final booster immunization (week 9). Polyprep® chromatography columns (Bio-Rad) were flushed with 20 mM phosphate buffer, pH 7.2, and packed with Gammabind Plus Sepharose (GE Healthcare). One column volume of 20 mM phosphate buffer, pH 7.2 was used to rinse the column material before loading the individual, 0.2 µm filtered mouse serum samples through the column material five times. Bound IgG was washed by rinsing the column with 100 mM Citric Acid (pH 4). After the washing procedure, mouse IgG was released by eluting with 100 mM Citric Acid (pH 2.7) in a neutralization buffer (to a final pH of 7.5 using 1 M Tris-HCl, pH 9). Eluted IgG samples were dialyzed (MWCO 12–14 kDa, Spectrum Labs) into PBS and concentrated with Vivaspin 6, MWCO 50 kDa. Finally, concentrations were measured by the IgG setting on the Nanodrop2000 (Thermo Scientific).
In vitro HCV neutralization assay
7 × 103 of HCV permissive Huh7.5 cells were plated per well in poly-D-lysine 96-well plates and incubated for 24 h. All neutralization assays were done with purified IgG instead of full serum to avoid non-IgG effects from mouse serum components. The following day, a dilution series of mouse-derived antibodies (at the highest dose of 0.5–1 mg/ml) was made using DMEM (Gibco) supplemented with 10% fetal calf serum (Sigma) and penicillin/streptomycin (Sigma) (full medium). This was incubated in a total volume of 10 μl with cell-culture infectious JFH1-based Core-NS2 recombinant HCVcc stocks, referred to by the isolate name of the envelope proteins: H77 (genotype 1a), TN (genotype 1a), J4 (genotype 1b), J6 (genotype 2a), T9 (genotype 2a), DBN (genotype 3a), ED43 (genotype 4a), SA13 (genotype 5a) and HK6a (genotype 6a)38,50,51,52,53, corresponding to a final read-out of 50–200 FFU per well. The virus/antibody mixes were, along with eight replicates of virus only, incubated for 1.5 h at 37 °C before addition of an additional 20 μl of full medium and incubation with Huh7.5 cells for 2.5 h at 37 °C in 5% CO2. Cells were washed, and fresh medium was added prior to incubation for a total infection time of 48 h before fixation. Following fixation, the cells were incubated with anti-mouse Fab fragments (Jackson Immuno Research) to block the Fc region of the mouse antibodies.
Cells were stained by overnight incubation with NS5A-specific antibody 9E10 in PBS buffer with BSK. Cells were washed three times in PBS buffer with 0.1%Tween20 and incubated 1 h at room temperature with secondary antibody Anti-mouse IgG, Horseradish Peroxidase (Amersham Biosciences) at a dilution of 1:2000 and visualized by DAB staining (VWR). FFUs were counted on an ImmunoSpot series 5 UV analyzer (CTL Europe GmbH) with customized software. The mean FFU count of 8 negative-control wells was subtracted from FFU counts in experimental wells. The data were normalized to 8 replicates of virus only and analyzed using three or four parameters curve fitting in GraphPad Prism 9.2.0, bottom set to 0, top set to 100.
Statistical analysis
All statistical analysis and graphs were prepared using the GraphPad Prism 9.2.0 software. The non-parametric, two tailed, Mann–Whitney Rank Sum Test unpaired comparison between immunization groups was used to test for statistical difference amongst different vaccination groups; statistical significance was defined as p < 0.05.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.

留言 (0)