Reagents
The following reagents were obtained as indicated: recombinant mouse IL-17A protein (BioLegend 576002), recombinant IFN-γ protein (PeproTech 315-05), lipopolysaccharide (List Biological Laboratories, Escherichia coli O111:B4), phorbol 12-myristate 13-acetate (PMA) (SIGMA P8139), ionomycin (I1957), and Golgi Plug (BD Bioscience, 555029).
Mice
A targeting vector for the Nfkbizfl/fl mice was constructed to delete exons 5, 6, and 7 of the Nfkbiz gene, which encode ankyrin repeats of IκBζ (Supplementary Fig. 1c). Genomic fragments of Nfkbiz were generated by PCR and cloned with loxP sequences and a Neor cassette flanked by FRT sequences as shown in Supplementary Fig. 1c. For the combinatorial use of CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR associated proteins) system, three sets of sgRNA oligos (5′-caccgaaggggtgcgggaacagtc-3′ and 5′-aaacgactgttcccgcaccccttc-3′ for pX335-G1, 5′-caccgagatagctgtctgagtacgc-3′ and 5′-aaacgcgtactcagacagctatctc-3′ for pX335-G2 and 5′-caccgtatcaatgtatcgttaaat-3′ and 5′-aaacatttaacgatacattgatac-3′ for pX-335-G3) were cloned into BbsI-digested pX335-U6-Chimeric_BB-CBh-hSpCas9n(D10A) plasmid (Addgene #42335). C57BL/6N-derived ES cell line, 6NK7 ES cells64 (2 × 106 cells) were co-electroporated with circular forms of 20 μg of the targeting vector and 10 μg each of pX335-G1, 2 and 3 using Gene Pulser XcellTM (Bio-Rad) and plated onto two 10 cm plates. After neomycin selection, ES clones in which single transgene was integrated into the Nfkbiz locus, were obtained. ES cells were aggregated with ICR morula as described.65 The chimeric mice were mated with C57BL/6 mice to establish a strain with a germline-transmitted locus. The resultant mice were bred with FLPe-expressing mice66 to delete Neor cassette flanked by FRT sequences. FLPe-expressing mice66 were obtained from Riken BioResource Research Center (RBRC01834, C57BL/6-Tg(CAG-flpe)36Ito/ItoRbrc). Vil1-Cre mice,32Lyz2Cre mice,67 and Nfkb1–/– mice68 were purchased from The Jackson Laboratory. Lyz2Cre mice were used for gene deletion in Paneth cells, as Lyz2 is specifically expressed in Paneth cells among IECs.60,69 All mice were maintained under SPF conditions in the animal facility at Toho University School of Medicine. The experimental protocols were approved by the Toho University Administrative Panel for Animal Care (22-54-413) and Recombinant DNA (22-54-410 and 22-53-442). Every effort was made to minimize the number and suffering of mice.
Fractionation of the small intestinal tissue
The terminal ilea isolated from wild-type mice (~10 cm) were incised longitudinally and the luminal contents were washed out in PBS. The tissue was treated with 10 mM EDTA in RPMI medium for 30 min at 37 °C. The dissociated cells were used as epithelial fraction after filtration with a 70 μm cell strainer (greiner bio-one 542070). The remaining tissue was washed in RPMI with vigorous shaking, and the middle portion (~1 cm) was used as lamina propria.
Animal disease models
In all disease model experiments, we used gender- and age-matched mice that had been co-housed since their birth. For DSS-induced colitis model, the disease was induced in mice by oral administration of 2.0% DSS (MW 36,000–50,000, MP Biomedicals Inc) ad libitum in drinking water for 5 days, and then normal drinking water was given in the following days. For induction of enteritis in the small intestine, an anti-CD3ε agonistic antibody (self-made, clone 145-2C11) was peritoneally injected to mice at day 0, 2, and 4 (1.0 mg/kg). In these two intestinal inflammation models, body weight was measured every 24 h, and the relative values to the initial body weight was shown. For induction of EAE model, mice were subcutaneously injected with the synthetic antigen peptide MOG35–55 (100 μg per mouse, Scrum Inc.) emulsified in complete Freund’s adjuvant without additional Mycobacterium tuberculosis H37RA. Pertussis toxin (500 ng per mouse, Calbiochem) was intraperitoneally injected to the mice on days 0 and 2. The severity of the disease was scored as follows: 0, no clinical signs; 1, tail limpness; 2, hind limb weakness; 3, hind limb paralysis; 4, fore limb weakness; 5, quadriplegia; 6, death.
In situ hybridization
Mice were perfused transcardially with 4% (w/v) paraformaldehyde (PFA) in phosphate-buffered saline (PBS, pH 7.4). The guts were removed, postfixed in 4% PFA at 4 °C overnight, followed by cryoprotection in 30% (w/v) sucrose for a day. The whole small intestine was cut longitudinally and the “Swiss roll” was made. The rolled tissue was embedded in the Surgipath (FSC22, Leica Biosystems), cryo-sectioned at the thickness of 20 μm, and then stored at −20 °C until use. Multicolour fluorescent in situ hybridization chain reaction was performed as previously described33 with some modifications using reagents from Nepagene Co. Ltd. (Chiba, Japan). Briefly, the section was treated with methanol for 10 min at room temperature, and then incubated in the hybridization solution containing 5× SSC, 10% dextran sulfate (MW. 500,000; Wako), 30% formamide, 0.1% Tween-20, 1× Denhardt’s solution and 50 μg/ml heparin for more than 5 min at 37 °C. After denaturing for 5 min at 95 °C, the DNA probe was diluted to the concentration of 10 nM with the hybridization solution, and applied onto the section. The section was covered by a piece of parafilm sheet, and incubated at 37 °C overnight in a moist chamber. After the section was sequentially washed with the solution containing 5× SSC, 30% formamide, and 0.1% Tween-20 for 10 min, followed by 5× SSC (10 min), the autofluorescence was quenched for an hour by the LED illuminator (TiYO, Nepagene, Japan). The section was incubated with the amplification buffer containing 8× SSC, 10% dextran sulfate, 0.2% Triton X-100, and 100 mM MgCl2 for more than 5 min, and then with the fluorophore-conjugated hairpin DNA (each 60 nM) for 2 h at 25 °C. Hairpin DNAs were snap-cooled (heated at 95 °C for 1 min, slowly cooled to 65 °C for 15 min, and then 25°C for 40 min) to form a hairpin structure before use. Finally, the section was stained with Hoechst 332342 (1 μg/ml) and mounted with anti-fade reagent (VECTASHIELD Mounting Medium, Vector Laboratories). The stained section was examined under a confocal laser microscope (A1R; Nikon, Tokyo, Japan). The nucleotide sequences of specific probes and hairpin DNAs were shown in Supplementary Table 1 and Supplementary Table 2, respectively.
In situ hybridization analysis for small intestinal organoids was conducted by RNAscope (Advanced Cell Diagnostics). After cultured on a chamber slide (Matsunami Glass, SCS-N08), organoids were fixed with formaldehyde (10%) and analyzed according to the manufacture’s protocol (https://www.cosmobio.co.jp/support/technology/document/ADC_Tech_Note_Mux_FL_CulturedCells_V2.pdf) using specific probe to Nfkbiz (806551), Lyz1 (415131-C2), Enpep (862211-C3), and Lgr5 (312171-C3). Pictures were obtained using the BZ-X700 All-in-one microscope (KEYENCE) or the confocal laser microscope LSM880 (Zeiss). Images were analyzed using BZ-X Analyzer (KEYENCE) or ZEN software (Zeiss).
Analysis on bacterial flora
DNA was isolated from the intestinal tissues and feces, and microbiota analysis was carried out as previously described.44 Briefly, the V4 region of the 16 S rRNA gene was amplified by PCR using barcoded dual-index primers that contain an Illumina adaptor and the V4-specific primers F515 and R806.70 The amplified fragments were pooled into a library, and the both ends of the fragments were sequenced using an Illumina MiSeq instrument. The obtained sequences were curated using Mothur (v.1.40.5), in which the sequences were binned into OTUs at >97% identity level and taxonomically assigned. After calculation of relative abundancies of individual OTUs and higher taxons, the indexes for α-diversity and β-diversity were obtained. Shannon evenness, which provides information on how much equal the abundancies of the OTUs are in a microbiome, was obtained by dividing Shannon index by natural logarithm of the total number of OTUs.
Measurement of IgA amount
The amount of IgA in feces was measured by enzyme-linked immunosorbent assay (ELISA) using Mouse IgA ELISA Kit (Bethyl laboratories, inc E99-103). The fecal IgA content was determined after normalization with the amount of total fecal protein measured using Protein Assay Dye Reagent (Bio-Rad 5000006).
Quantitative polymerase chain reaction (qPCR)
Quantitative PCR (qPCR) was conducted using FAST SYBRTM Green Master Mix (Applied biosystems, 4385614) on QuantStudio 3 real-time PCR system (Applied biosystems). The sequences of qPCR primers used were listed in Supplementary Table 3.
Gene expression analysis by reverse transcription–qPCR (RT-qPCR)
Total RNA was isolated using SepasolⓇ-RNA Super G (nacalai tesque). For extraction of RNA from the intestinal tissues, the tissue fragment was ground with a Zirconia bead (TOMY, ZB-50) in SepasolⓇ-RNA Super G using Micro Smash MS-100R (TOMY Digital Biology). The obtained RNA was reverse-transcribed to complementary DNA (cDNA) using ReverTra AceⓇ qPCR RT kit (TOYOBO, FSQ-101). cDNA was used as a template in qPCR. The relative expression level of every gene was determined after normalization to that of the house-keeping gene Hprt.
Measurement of the bacteria contents in the intestinal tissues and feces
DNA was extracted from mouse intestinal tissues and feces using QIAamp DNA Mini Kit (QIAGEN, 51304) and QIAamp Fast DNA Stool Mini Kit (QIAGEN, 51604), respectively. The obtained DNA was used as a template of qPCR. The content of bacteria was determined after normalization to the amount of total eubacteria or Actb.
Chromatin immunoprecipitation (ChIP)
ChIP assay was essentially performed as previously described.52 Cells were fixed with formaldehyde (1.0%) for 10 min at room temperature, and sonicated. The fragmented chromatin was subjected to immunoprecipitation using anti-IκBζ antibody,31 anti-NF-κB p65 antibody (Santa Cruz, sc-372), or rabbit control IgG (GeneTex, GTX35035). After reversal of the cross-linking, the precipitated DNA was analyzed by qPCR. The association of the transcription regulator with the target site was given after normalization to the amount of input DNA.
Immunoblotting
Immunoblotting was performed according to the standard protocol using antibodies against total-STAT1 (Cell signaling Technologies 9172), phospho-STAT1 (S727) (Cell signaling 8826S), and α-tubulin (Sigma-Aldrich T5168).
Microarray chip analysis
Total RNA was prepared from the ilea of two pairs of Nfkbizfl/flVil1-Cre mice and co-housed gender-matched control (Nfkbizfl/fl) mice. Labeling of cRNA was performed using Agilent Low Input Quick Amp Labeling Kit following the manufacture’s instructions (Agilent Technologies). Briefly, after total RNA was reverse-transcribed to double-stranded cDNA using a poly(dT)-T7 promoter primer, the resultant cDNA was used as a template for in vitro transcription in the presence of Cyanine 3 (Cy3)-CTP. The Cy3-labeled cRNA was fragmented, and hybridized onto Agilent SurePrint G3 Mouse GE v3 8 × 60 K Microarray (Design ID: 074809). After washed, the microarray was scanned using an Agilent SureScan Microarray Scanner (G4900DA). Intensity values of each scanned feature were quantified using Agilent feature extraction software version 12.1.1.1, which performs background subtractions. We employed features that were fragged as no errors (Detected flags), and excluded features that were not positive, not significant, not uniform, not above background, saturated, and population outliers (Not Detected and Compromised flags). Quantile normalization was performed using Agilent GeneSpring software 14.9.1.
Flow cytometric analysis on the lamina propria lymphocytes in the small intestine
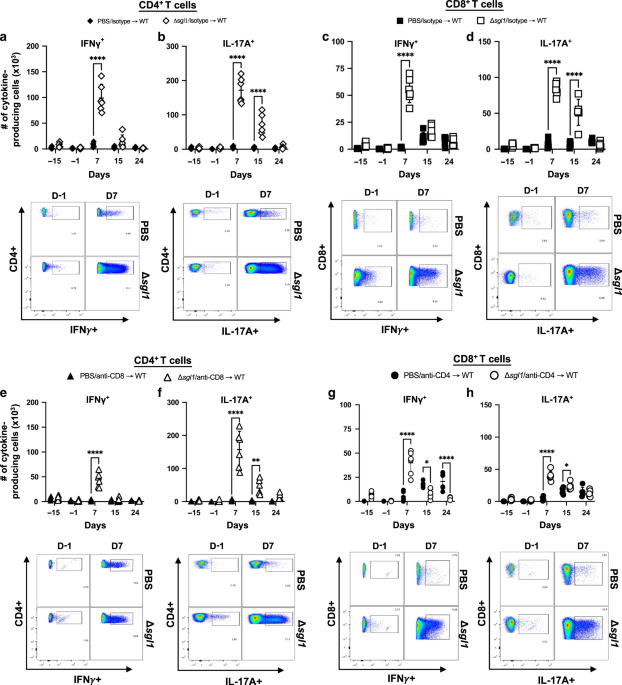
Non-epithelial cells were prepared from the lamina propria of the small intestine as described previously.71 After epithelium was removed by incubation in 2 mM EDTA, the remaining intestinal tissue was chopped into small pieces, and digested with collagenase (0.5 mg/ml, Wako). The obtained cell suspension was suspended in 40% Percoll solution (GE Healthcare) and loaded on a 80% Percoll solution. After centrifugation for 20 min at 880 × g at room temperature, cells at interface between the two Percoll solutions were harvested. After stimulation for 4 h with PMA (50 ng/ml) and ionomycin (500 ng/ml) in the presence of GolgiPlug (1:1000 dilution), cell surface was stained with FVD506 (eBioscience 65-0866-14), APC/Cy7-labeled anti-CD45.2 antibody (BioLegend 109824, clone 104), APC-labeled anti-CD4 antibody (TONBO 20-0042, clone RM4-5), PE/Cy7-labeled TCRβ antibody (TONBO 60-5961, clone H57-597). The cells were fixed with Intracellular Fixation Buffer (eBioscience 00-8222) and then perforated in Permeabilization Buffer (eBioscience 00-8333). Intracellular cytokines were stained with PE-labeled anti-IL-17A (BioLegend 506904, clone TC11-18H10) and FITC-labeled anti-IFN-γ (BioLegend 505806, clone XMG1.2) in Permeabilization Buffer, and analyzed using LSR FortessaTM X-20 Cell Analyzer (BD Bioscience) and FlowJo (BD Bioscience).
Preparation of the small intestinal organoid culture
Preparation and maintenance of the small intestinal organoid culture were carried out according to the instruction of Intestinal Epithelial Organoid Culture with IntestiCultTM Organoid Growth Medium (Mouse) (STEM Cell technologies 06005). For dissociation of crypts from the intestinal tissue, the jejunum was cut into small fragments, extensively washed with cold PBS, and incubated at 4 °C in 10 mM EDTA. The isolated crypts were suspended in Matrigel (Corning 356231) and cultured in the complete medium (STEM Cell technologies 06005). For inoculation, organoid-containing Matrigel was dissociated with Gentle Cell Dissociation Reagent (STEM Cell technologies 07174), and the organoids were washed with DMEM/F-12 with 15 mM HEPES (STEM Cell technologies 36254), and re-suspended in Matrigel for re-plating. For the experiments of recovery from IFN-γ-induced Paneth cell death, the IFN-γ-treated organoids were washed twice, and re-cultured in fresh media under IFN-γ-free conditions
Preparation of bone marrow-derived macrophages
Bone marrow-derived macrophages were prepared as described previously,52 and maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal calf serum, penicillin (100 units/ml), and streptomycin (100 μg/ml).
Histological analysis
The tissue from the intestine or the spinal cord was fixed in formaldehyde (10%) overnight at room temperature, and embedded in paraffin blocks. The paraffin-embedded section was used for H&E staining or immunohistochemical analysis. For immunohistochemical analysis, paraffin-embedded section was autoclaved in citrate buffer (LSI Medicine, RM-102C) for retrieval of the antigens, and then stained with anti-Lysozyme antibody (abcam, ab108508). After treatment with HRP-conjugated secondary antibody using ImmPRESS VR Polymer HRP Anti-Rabbit IgG Reagent (Vector Laboratories, MP-6401), the distribution of Lysozyme was visualized with 3-3′-diaminobenzidine (nacalai tesque, 11009-41). The section of the spinal cord was stained with Luxol Fast Blue (Muto Pure Chemicals, 4100-1) followed by H&E staining. Small intestinal organoids cultured on a coverslip were fixed with PFA (4%), and permeabilized in a buffer containing normal goat serum (5%) and Triton X-100 (0.5%) in BSA (2%). For fluorescent analyses, sections were stained with fluorescein-labeled Ulex Europaeus agglutinin 1 (UEA-1) (Vector laboratories, FL-1061-2), Alexa 647-labeled anti-E-Cadherin antibody (BD, 560062 clone 36/E-Cadherin), and Hoechst 33258 (Nacalai, 04928-92). Pictures for the images were obtained using the BZ-X700 All-in-one microscope (KEYENCE). Images of organoids were processed using acquisition of Z-stacks followed by haze reduction function.
RNA sequencing (RNA-seq)
The 5′ RNA sequencing was performed by ImmunoGeneTeqs, Inc. (Chiba, Japan). PolyA+ RNA was isolated using Dynabeads M-270 Streptavidin (Thermo Fisher Scientific) and biotin-3′ WTA-EcoP-dT25, according to the previous analysis (GSE110711) with some modifications. After reverse transcription and template switching, the cDNA was amplified and subjected to fragmentation/end-repair/polyA-tailing/ligation using NEBNext Ultra II FS DNA Library Prep Kit for Illumina (New England Biolabs). Barcoded libraries (~300 bp) were obtained by PCR using NEBNext Ultra IIQ5 Master Mix (New England Biolabs), and sequenced on the Illumina Novaseq 6000 S4 flowcell (Illumina). After adaptor trimming of sequencing data in single-end fastq files using cutadapt 2.10, the reads were mapped to reference RNA (mRNA and ncRNA of GRCm38 release 101) with bowtie 2-2.3.4.2. The reads in each gene were counted using awk, sort and uniq -c commands. The count data was summarized, and the expression table was full-outer joined by gene symbols using Microsoft R open-3.5.3 and dplyr-1.0.0 package.
Injection of anti-IFN-γ antibody
Co-housed Nfkbizfl/flVil1-Cre mice were intraperitoneally injected with an isotype control antibody (ichorbio ICH2246) or an anti-IFN-γ antagonistic antibody (clone R4-6A2) (15 mg/kg) every 3 days. Twenty-four hours after the last injection, mice were sacrificed to remove the intestinal tissues.
Assay for transposase-accessible chromatin using sequencing (ATAC-seq)
The cryopreserved jejunum was sent to Active Motif for ATAC-seq experiments. The nuclei isolated from the tissue were tagmented (fragmented and tagged with sequencing adaptors) by hyperactive Tn5 transposase as described previously72 with some modifications73 using the reagents in Nextera Library Prep Kit (Illumina). After amplification with ten cycles of PCR, the resultant DNA was sequenced with PE42 sequencing on the NextSeq 500 sequencer (Illumin). Sequence reads were aligned using the BWA algorithm,74 and peaks in the histograms were identified using the MACS 2.1.0 algorithm at a cutoff of p value 1 × 107. Signal maps and peak locations were analyzed using Active Motifs proprietary analysis program, and reads counted in all merged peak regions were compared using DeSeq2.75
Statistical analysis
For statistical analysis of mouse experiments, pooled data from independent experiments are presented unless otherwise indicated. All statistical analyses were conducted using Prism software (version 9.2.0) and the details of each experiment are shown at the end of the respective figure legend. Significance of the statistics is defined as *p value < 0.05, **p value < 0.01, ***p value < 0.001, ****p value < 0.0001.


留言 (0)