
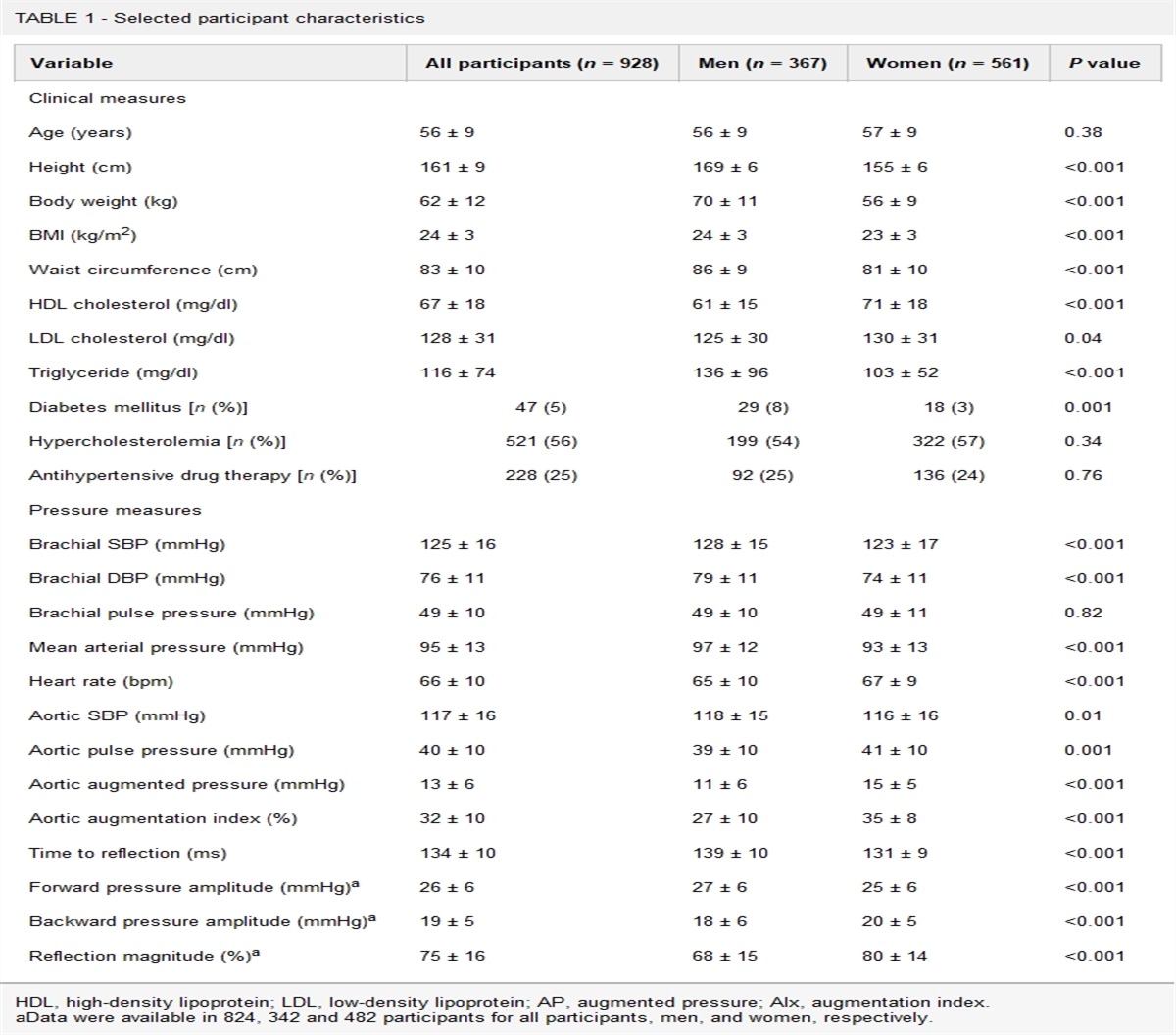
記住我

Pulmonary hypertension (PH) is an inappropriate elevation of mean pulmonary arterial pressure (mPAP ≥25 mmHg) attributable to known or unknown causes [1]. By expert consensus, the World Health Organization has classified PH into five groups (Table 1). Over the last 10 years, multicenter registries and clinical trials regarding the increased mortality risk in PH patients with mPAP <25 mmHg have prompted a reconsideration of these thresholds [2,3]. The normal mPAP range is approx. 14 ± 3.3 mmHg, with a normal upper limit of approx. 20 mmHg. The recent World Symposium on PH thus updated new PH hemodynamic thresholds to define precapillary PH, including the mPAP value >20 mmHg, the pulmonary artery wedge pressure (PCWP) value ≤15 mmHg, and the pulmonary vascular resistance (PVR) value of ≥3 Wood units [4]. One subtype of PH is pulmonary arterial hypertension (PAH). The pathobiology of PAH involves endothelial cell dysfunction and proliferation and the migration of pulmonary arterial smooth muscle cells (PASMCs), leading to severe occlusive pulmonary arterial remodeling [5]. Platelet-derived growth factor (PDGF)-BB, which is an essential mitogen and chemoattractant for PASMCs, has been implicated in these complicated cellular processes [6]. Laboratory studies demonstrated that the inhibition of PDGF receptor-B (PDGF-RB) by the inhibitor ST1571 impaired the proliferation of PASMCs to reverse progressive pulmonary vascular changes [6]. Over the past two decades, despite advancements in therapy, PAH remains a devastating disease that markedly reduces patients’ survival [1]. Unfortunately, no potential molecular therapies are currently in clinical use that adequately cure or reverse PAH, which would lead to improvements in the symptoms and mortality of PAH patients [7].
TABLE 1 - Clinical classification of pulmonary hypertension from the Fifth World Symposium on Pulmonary Hypertension (Nice, France) Group Pathogenesis/diagnosis Etiology Group 1 Pulmonary hypertension is a clinical condition defined as a mean of pulmonary artery pressure ≥25 mmHg/pulmonary capillary wedge pressure ≤15 mmHg, which is identified by the presence of pre-capillary pulmonary hypertension without other causes of pre-capillary pulmonary hypertension, such as PH due to lung disease, CTEPH, or another rare disease Idiopathic Familial/heritable Mutation genes in blow: ACVRL1BMPR2 (TGF-βR superfamily member) CAV1EIF2AK4 Endolin (TGF-βR superfamily member)KCNK3/TASK1SMAD9 (TGF-β superfamily member) unknown related to: 1. Congenital heart disease2. Connective tissues disease3. Portal hypertension (liver)4. HIV infection5. Schistosomiasis Drug/toxin-caused PCHNB PCH and/or PVOD Group 2 PH caused by left ventricular dysfunction, which is identified by the passive backward transmission of the pressure elevation (post-capillary PH) 1. SHF and/or DHF2. Valvular disease3. Cardiomyopathies with congenital/acquired left ventricular inflow /outflow tract obstruction Group 3 PH caused by lung and/or hypoxia, which is the result of hypoxic vasoconstriction 1. COPD2. ILD3. Developmental lung disease4. Sleep-disordered breathing5. Other pulmonary diseases with complex restrictive and obstructive pattern Group 4 CTEPH caused by chronic mechanical obstruction of distal and central pulmonary arteries by organized thrombi Group 5 PH caused by uncertain multi-factorial mechanisms 1. Chronic hemolytic anemia2. Systemic diseases (e.g. pulmonary histiocytosis, sarcoidosis, etc.)3. Myeloproliferative diseases, splenectomy4. Metabolic diseases (e.g. thyroid disorder, glycogen storage disease, etc.)5. Other diseasesACVRL1/ALK1, activin-receptor-like kinase-1; BMPR2, bone morphogenetic protein receptor type 2; CAV1, caveolin-1; COPD, chronic obstructive pulmonary disease; CTEPH, chronic thromboembolic pulmonary hypertension; DHF, diastolic heart failure; EIF2AK4, eukaryotic translation initiation factor 2 alpha kinase 4; ILD, interstitial lung disease; KCNK3/TASK-1, a second K2P channel; PCH, pulmonary capillary hemangiomatosis; PCHNB, persistent capillary hypertension of the newborn; PVOD, pulmonary veno-occlusive disease; SHF, systolic heart failure.
A member of the protein tyrosine phosphatase family, i.e. protein tyrosine phosphatase receptor type D (PTPRD), has functions including the regulation of cell growth and differentiation [8]. PTPRD gene was initially described as a tumor suppressor in several carcinoma cell lines [9]. Later studies have focused on the role of PTPRD in cancer and neurologic disease. Microarray data from public databases were re-analyzed, identifying PTPRD as a candidate gene for the radiation resistance observed in patients with nasopharyngeal carcinoma [10]. PTPRD has been also targeted for medications that are intended to combat cocaine use disorders [11]. A PTPRD genetic variant appears to be associated with progression to diabetes in Han Chinese individuals, most likely through increased insulin resistance [12]. A recent study demonstrated that PTPRD gene variants can interact with maternal perceived stress to affect the risk of spontaneous preterm birth [13]. It was also indicated that PTPRD might be likely to correlate with PAH. PTPRD has been observed to be down-regulated by PDGF-BB in PASMCs, suggesting its promising biological activity as a novel therapeutic candidate in the management of PAH in humans and animals. However, the role of PTPRD gene in the development of PAH and its underlying mechanism have not been investigated, even in an experimental animal model.
In this issue of the Journal of Hypertension, Xu et al. present intriguing results regarding PTPRD gene methylation- and silencing-induced PAH in a rat model's response to hypoxia, hypoxia-sugen, and monocrotaline [14], and they provide two pieces of new information. First, they demonstrated that PTPRD silencing-mediated PDGF-RB/PLCγ1 signaling activation is responsible for the PASMC migration and severe pulmonary arterial remodeling that occur in the development of PAH. Second, they indicated that the epigenetic regulation of PTPRD by PDGF-BB might facilitate PASMCs’ function and pulmonary artery remodeling in PAH development in an animal model. Increasing evidence shows that PTPRD plays a vital role in progressive cancers and diabetic diseases [9–12,15]. To the best of our knowledge, the Xu et al. study is the first to explore the role of PTPRD in PASMC function during the development of PAH in rats [14]. In the rats, PTPRD deletion not only accelerated pulmonary artery neointimal formation and remodeling; it also increased the right ventricular systolic pressure (RVSP) and right ventricular hypertrophy index (RVHI) [14], suggesting that PTPRD is an important mediator of hypoxia-induced pulmonary artery and right ventricular responses. This notion was further supported by the data of two additional in-vivo analyses revealing that the disruption of PTPRD also exerted a harmful effect on pulmonary artery morphological changes as well as the RVSP and RVHI in rats in response to hypoxia-sugen and monocrotaline [14]. Cell motility is known to be a complex and dynamic process involving the localization of a microtubule-organizing center. In vitro, PTPRD silencing stimulated the migration of cultured rat PASMCs via the modulation of cell focal adhesion and the cytoskeleton, whereas it had no effect on proliferation [14]. PTPRD has been shown to dephosphorylate PDGF-RB at a Tyr1009 site (PDGF-RBTyr1009) in the murine cortex, leading to chemotaxis and cell migration [16,17]. The present results reported by Xu et al. demonstrate that PTPRD knockdown caused an upregulation of PDGF-RBTyr1009 in PASMCs. Moreover, a PDGF-RBTyr1009-phosphorylation-mediated activation of phospholipase C gamma 1 (PLCγ1) can promote PTPRD-silenced PASMC migration. Because PLCγ1 can bind to the phosphorylated Tyr1009 site on PDGF-RB, and subsequently be phosphorylated at Tyr783 in order to be activated [18], the silencing of PTPRD appears to modulate PASMC migration via a PDGF-RB/PLCγ1 axis and thus lead to pulmonary hypertension (Fig. 1).
 FIGURE 1:
FIGURE 1: Proposed mechanisms of PTPRD's epigenetic and genetic modification-mediated pulmonary artery remodeling leading to PAH. DNMT1, DNA methyltransferase; PA, pulmonary artery; PAH, pulmonary arterial hypertension; PASMC, pulmonary artery smooth muscle cell; PDGF-BB, platelet-derived growth factor-BB; PDGF-RB, platelet-derived growth factor receptor B; PLCγ, phospholipase C gamma 1; PTPRD, protein tyrosine phosphatase receptor type D; RVHI, right ventricular hypertrophic index; RVSP, right ventricular systolic pressure.
Another possible mechanism by which PDGF-BB epigenetically modulates PTPRD was also investigated by Xu et al.[14]. PTPRD, which possesses a canonical promoter CpG island across a transcription STAT site, was reported to be silenced via promoter hypermethylation in proliferative diseases [19]. In the Xu et al. study in this issue, PDGF-BB dramatically downregulated the expression of PTPRD mRNA in human PASMCs, and this effect was restored by pretreatment with the DNA methyltransferase inhibitor 5-Aza-dC [14], indicating that PDGF-BB negatively regulates PTPRD by DNA methylation of the CpG islands of the PTPRD promoter. Among the members of the DNA methyltransferase (DNMT) family, DNMT1 acts as a maintenance DNMT, whereas DNMT3A and DNMT3B are de novo DNMTs [20]. The result obtained by Xu et al. also shows that DNMT1 was sensitive to a PDGF-BB stimulator. Collectively, these findings suggest that PTPRD promoter methylation by PDGF-BB might facilitate pulmonary artery remodeling via the modulation of PASMCs’ functions in this experimental PAH model (Fig. 1).
Finally, Xu et al. examined the impact of hypoxia on PTPRD expression in vitro and in vivo. They observed that hypoxia suppressed PTPRD gene expression in rat cultured PASMCs. In vivo, chronic hypoxia also resulted in severe pulmonary arterial remodeling and increased RVSP and RVHI values, accompanied by reductions of PTPRD mRNA and protein [14]. Subsequently, Xu et al. observed not only a downregulation of PTPRD expression but also elevations of RVSP and RVHI [14]. Consistently, the data of patients with idiopathic and heritable PAH have confirmed that their PTPRD expression levels were significantly decreased in blood and/or pulmonary arteries [14], indicating that PTPRD might serve as novel biomarker as well as a potential therapeutic target for patients with PAH.
Of course, the results presented by Xu et al. are only the beginning of this exciting new direction in the therapeutic control of experimental PAH by epigenetic and genetic modifications of PTPRD [14]. Given the potent multifunctional effects of PTPRD, which is known as a suppressor of proliferative diseases, several issues must be investigated. In particular, it must be investigated whether it is possible to genetically modify PTPRD gene or PTPRD overexpression in order to produce a pulmonary vasculoprotective effect in patients with arteriosclerosis- and/or thrombosis-based PAH. Moreover, there is a profound need to further our pathophysiologic knowledge to develop PTPRD-based novel molecular therapeutic drugs for preclinical and clinical challenges (e.g., small molecular compounds, PTPRD agonists/antagonists, small polypeptides, small RNA or DNA nucleotides, etc.).
In conclusion, in light of the findings suggesting that PTPRD could become a novel molecular target for the management of experimental PAH, further studies will shed light on whether a PTPRD modification-mediated mitigation of PASMC adhesion and migration could contribute to the reversal of pulmonary artery remodeling and the development of PAH in humans. Addressing the above-mentioned questions will also better guide the clinical evaluation and care of patients with PAH.
ACKNOWLEDGEMENTSFunding: This work was supported in part by grants from the National Natural Science Foundation of China (nos. 81770485 and 81560240).
Conflicts of interestThere are no conflicts of interest.
REFERENCES 1. Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 2014; 115:189–202. 2. Douschan P, Kovacs G, Avian A, Foris V, Gruber F, Olschewski A, et al. Mild elevation of pulmonary arterial pressure as a predictor of mortality. Am J Respir Crit Care Med 2018; 197:509–516. 3. Maron BA, Hess E, Maddox TM, Opotowsky AR, Tedford RJ, Lahm T, et al. Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the Veterans Affairs clinical assessment, reporting, and tracking program. Circulation 2016; 133:1240–1248. 4. Ruopp NF, Farber HW. The new world symposium on pulmonary hypertension guidelines: should twenty-one be the new twenty-five? Circulation 2019; 140:1134–1136. 5. Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest 2005; 115:2691–2694. 6. Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 2005; 115:2811–2821. 7. Cheng X, Narisawa M, Jin E, Li X. Alpha-solanine as potential therapeutic target in pulmonary artery hypertension. J Hypertens 2017; 35:2377–2379. 8. Uhl GR, Martinez MJ. PTPRD: neurobiology, genetics, and initial pharmacology of a pleiotropic contributor to brain phenotypes. Ann NY Acad Sci 2019; 1451:112–129. 9. Nair P, De Preter K, Vandesompele J, Speleman F, Stallings RL. Aberrant splicing of the PTPRD gene mimics microdeletions identified at this locus in neuroblastomas. Genes Chromosomes Cancer 2008; 47:197–202. 10. Lin Y, Zhou X, Yang K, Chen Y, Wang L, Luo W, et al. Protein tyrosine phosphatase receptor type D gene promotes radiosensitivity via STAT3 dephosphorylation in nasopharyngeal carcinoma. Oncogene 2021; 40:3101–3117. 11. Uhl GR, Martinez MJ, Paik P, Sulima A, Bi GH, Iyer MR, et al. Cocaine reward is reduced by decreased expression of receptor-type protein tyrosine phosphatase D (PTPRD) and by a novel PTPRD antagonist. Proc Natl Acad Sci USA 2018; 115:11597–11602. 12. Chang YC, Chiu YF, Liu PH, Shih KC, Lin MW, Sheu WH, et al. Replication of genome-wide association signals of type 2 diabetes in Han Chinese in a prospective cohort. Clin Endocrinol (Oxf) 2012; 76:365–372. 13. Hong X, Surkan PJ, Zhang B, Keiser A, Ji Y, Ji H, et al. Genome-wide association study identifies a novel maternal gene × stress interaction associated with spontaneous preterm birth. Pediatr Res 2021; 89:1549–1556. 14. Qin X, Fang E, Narisawa M, Cheng XW. Alternating P wave morphology. Circulation 2019; 139:1225–1227. 15. Purdie KJ, Harwood CA, Gulati A, Chaplin T, Lambert SR, Cerio R, et al. Single nucleotide polymorphism array analysis defines a specific genetic fingerprint for well-differentiated cutaneous sccs. J Invest Dermatol 2009; 129:1562–1568. 16. Tomita H, Cornejo F, Aranda-Pino B, Woodard CL, Rioseco CC, Neel BG, et al. The protein tyrosine phosphatase receptor delta regulates developmental neurogenesis. Cell Rep 2020; 30:215–228 e215. 17. Ronnstrand L, Arvidsson AK, Kallin A, Rorsman C, Hellman U, Engstrom U, et al. SHP-2 binds to Tyr763 and Tyr1009 in the PDGF β-receptor and mediates PDGF-induced activation of the Ras/MAP kinase pathway and chemotaxis. Oncogene 1999; 18:3696–3702. 18. Valius M, Bazenet C, Kazlauskas A. Tyrosines 1021 and 1009 are phosphorylation sites in the carboxy terminus of the platelet-derived growth factor receptor beta subunit and are required for binding of phospholipase C gamma and a 64-kilodalton protein, respectively. Mol Cell Biol 1993; 13:133–143. 19. Chen Y, Li B, Huang W, Gao D, Liang Z. Efficient and reproducible CH3NH3PbI(3 − x)(SCN)x perovskite based planar solar cells. Chem Commun (Camb) 2015; 51:11997–11999. 20. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6–21.
留言 (0)