
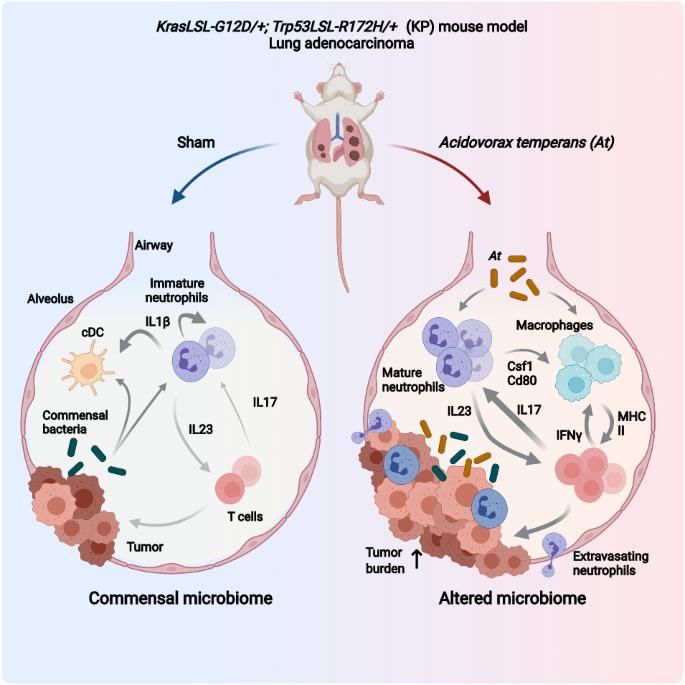
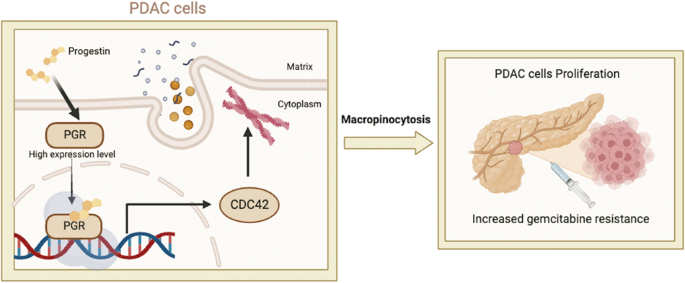
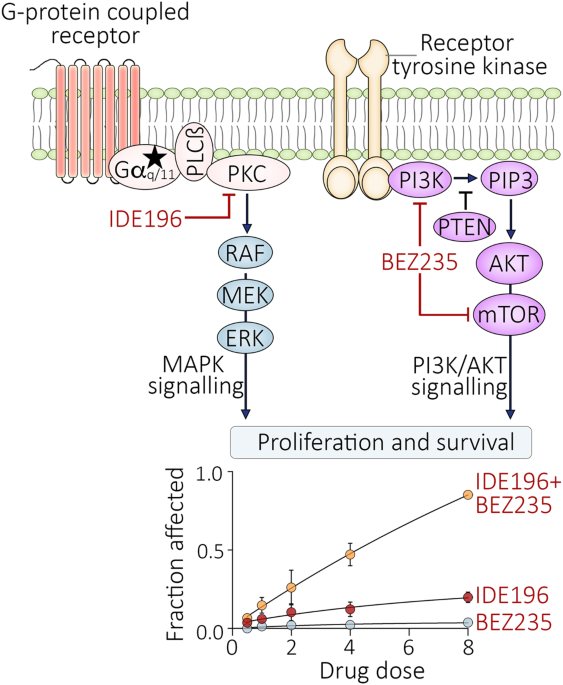
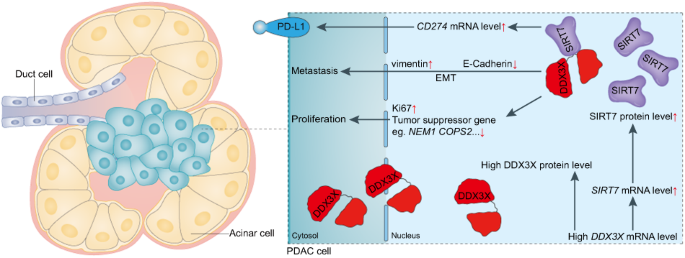
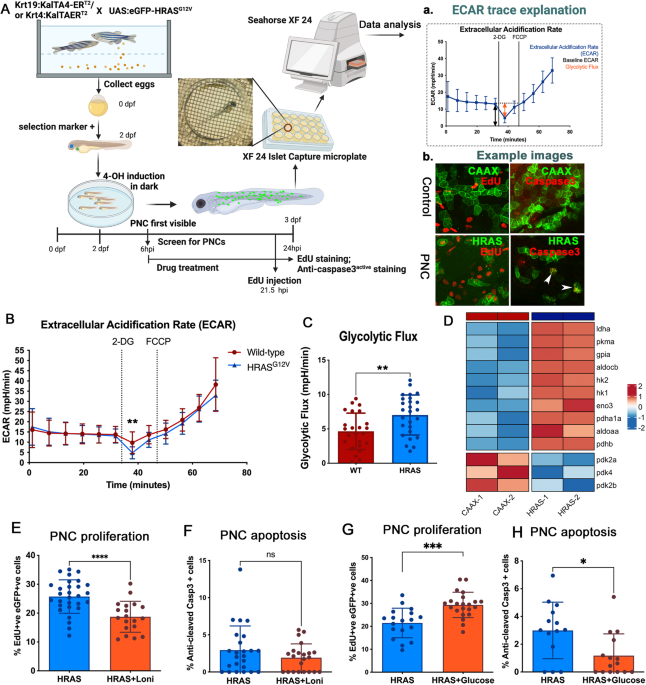
記住我


We collected genomic DNA from Brca1+/− mice at different developmental time points from embryo to adulthood. We performed whole-genome sequencing for each DNA sample, analyzed genomic sequences to search for the evidence of genome instability represented by SV, Indel and CNV, and compared the data between different time points. We also generated Brca1+/− Trp53+/− mice, collected and sequenced the DNA at the same time points, and compared the variation data between Brca1+/− mice and Brca1+/− Trp53+/− mice (Fig. S1).
Genome instability appeared at embryonic stage and dynamically changedTo monitor genome stability across the developmental stages, we performed whole-genome sequencing in the DNA samples in Brca1+/− mice from 10.5 and 16.5 embryonic days to adulthood at 1st, 4th, 8th and 12th months after birth. We performed bioinformatics data analysis to identify genetic changes in the sequence data in each DNA sample. We observed that SVs, CNVs, and indels were already present at the 10.5 embryonic day in the Brca1+/− mice, with multiple clusters present in different chromosomes (Fig. 1a, Table S2, S3). The variations changed dynamically, some were intensified, others were diminished and/or intensified again along developmental process. For example, the SV cluster chr.9: 70628040-79756364 appeared at 16.5 embryonic days, intensified at 4th months then nearly disappeared afterwards; the SV cluster chr2: 38128829–41439173 appeared at 10.5 embryonic days, intensified at 16.5 embryonic days, then disappeared at the 1st and 4th month but appeared again at the 12th month (Fig. 1b). At the gene level, the mutations affecting Hist1h2bc, P7, Vamp3, Cdk6, Nj1, Msh5he, Tirap, Tfrsf21, Marf1 were only present at specific developmental time points, whereas the mutations affecting Ccnd3, Fgfr2 appeared at early time, disappeared at the 8th month, and re-appeared at the latter time (Fig. 2).
Fig. 1: Monitoring genome instability across developmental stages.
Brca1+/− mice in 10.5E, 16.5E, 1M, 4M, 8M and 12M (n = 2 in each group) were tested. a SVs, Indels, and CNVs at different time points. Data from 2 mice in each group were combined and divided by 2 to obtain the average value. X-axis: the time points. Y-axis: the mutation frequency normalized by the median. b Representative Circos plots showing the variation at different time points. Outer: indels; middle: CNVs (red: gain; blue: loss); inner: SVs (red: gene affected; black: no gene affected).
Fig. 2: Dynamic changes of functionally important genes disrupted by SVs from embryonic towards adulthood stages.
Heatmap showed the functionally important genes affected by SVs in Brca1+/− mice. Mutation frequency in the genes at each time point was represented in color gradient ranging from blue to red. It shows that certain disruptions generated at early developmental stage were constantly present across the entire developmental stage, whereas others were only present at given time point(s). Brca1 mutation (at the left) was present at each time point.
We compared the mutation distribution and identified multiple mutation hot-spots of SV, CNVs and Indels across the genomes, as represented by the four clusters of chr4: 139320925-151922486, chr5: 3152512-8342821, chr11: 9607557–107346908, and chr13: 11440505-3139770 (Fig. 1b, Fig. 3). This pattern was not present in wild-type control Brca1+/+ mice (Fig. S2, Table S4), highlighting that the changes in the Brca1-knockout mice were unlikely derived from background variation.
Fig. 3: Variant distribution across different chromosomes.
a Distribution of SV breakpoints in Brca1+/− mice. Red dot represents the frequency of breakpoint occurrence in the corresponding site. Green circle marks the clustered region. Variant distribution of Brca1+/− mice in the four major SV clusters detailed in (b–e). (b) chr4: 139320925–151922486; (c) chr5 3152512-8342821; (d) chr11: 96075557-107346908; (e) chr13: 111440505-3139770. Each cluster shows SVs [purple: break end (BND); orange: inversion; red: duplication; blue: deletion)], CNVs (blue: loss; red: gain) and Indels (yellow: insertion; blue: deletion). The curves in SVs refer to their interaction with other genomic regions. Chromosomal bands are indicated in each cluster. Representative genes affected are listed at the bottom of each cluster.
Genome instability targeted repetitive sequences and fragile sitesWe analyzed the sequences at the SV breakpoint sites in Brca1+/− mice to determine the type of sequences susceptible to the damage. The results showed that 54% of SV break sites were located at repetitive sequences of simple repeats, LINE/L1, and LTR/ERVK. The rate was much higher than the 45% of the repetitive sequences in the mouse genome (Fig. 4a) [18]. Multiple chromosomal fragile sites including Astn2, Il1rapl1, Rev3l, Thsd7a and Wwox were also present at the breakpoint sites (Table S5) [19]. The results indicated that repetitive sequences and fragile sites were vulnerably attacked by the heterozygotic Brca1 mutation-caused genome instability.
Fig. 4: Repetitive sequences and break repair by error-prone non-homologous repair pathways.
a Repetitive sequence classification identified at SV breakpoints sites in Brca1+/− mice. b Number of SV breakpoints repaired by error-prone non-homologous pathways of NHEJ, MMEJ, and SSA. It shows that NHEJ contributed the majority of the error-prone repairs.
Genome instability promoted the use of error-prone no-homologous repair pathwaysBrca1 mutation damages the error-free homologous recombination (HR) pathway but promotes the use of error-prone non-homologous end joining (NHEJ) pathways to repair double-strand DNA breaks [20]. We analyzed micro-homogenous bases at both ends of SV breakpoint sites to assess the effects of heterozygotic Brca1 mutation on non-homologous repair pathways. Based on the presence of micro-homologous bases (NHEJ 1–5 bp, MMEJ (microhomology-mediated end joining) 6–25 bp, and SSA (single-strand annealing) > 25 bp) [21,22,23], we identified 569 repaired double-strand break events by the non-homologous repair pathways, including 492 in NHEJ, 75 in MMEJ, and 2 in SSA (Fig. 4b, Table S6). The enrichment of NHEJ, MMEJ, and SSA-repaired damage implied that the defects in error-free homologous recombination function caused by Brca1 mutation indeed promoted the use of error-prone non-homologous DNA repair pathways to repair the damaged double-stranded DNA, which further enhanced Brca1 mutation-caused genome instability.
Genome instability affected functionally important genes and pathwaysOverall, the genome instability by deletion, duplication, translocation, inversion caused by SVs, indels, and CNVs at different developmental stages affected over 2,300 genes in the Brca1+/− mice genomes. Many of these affected genes are functionally important involving in oncogenesis, tumor suppression, DNA damage repair, and immune function (Table S7a, S7b). For example, Msh5 is involved in DNA mismatch repair and meiotic recombination [24]. A deletion between Msh5 and 1700031A10Rik at the 4th month formed Msh5-1700031A10Rik out-of-frame fusion; Samd9 is a tumor suppressor involving in cell proliferation and innate immune response to viral infection [25]. A duplication in Samd9 occurred at 16.5 embryonic days; Aldoa plays a role in glycolysis and gluconeogenesis [26]. A t(7:12) translocation at the 4th month formed an out-of-frame Aldoa-Aldoart2 fusion; Rere is involved in apoptosis. An inversion at 16.5 embryonic day disrupted Rere structure (Fig. 5); Rad51b is critical for double-stranded DNA break repair in the homologous recombination pathway [27]. A t(12:14) translocation at the 4th month formed Rad51b-Fbxo34 fusion; Ccnd3 regulates G1/S transition and is frequently dysregulated in many cancer types [28]. A t(4:17) translocation at 16.5 embryonic day disrupted Ccnd3; Fgfr2 has tyrosine kinase activity and is frequently mutated in cancer [29]. A t(7:11) translocation at the 4th month disrupted Fgfr2; Hdac9 regulates histone deacetylation [30]. A t(7:12) translocation at the 4th month formed Sptbn4-Hdac9 fusion; Elf1 is a transcriptional factor [31]. A frameshift insertion at the 4th month disrupted Elf1; Pik3cd phosphorylates inositol lipids in immune response [32]. A t(4:8) translocation at the 1st month formed Pik3cd-Wwox fusion; B2m is an MHC class I protein playing key roles in antigen presentation [33]. Inversion of the B2m at the 4th month disrupted B2m. Many mutations were located in non-coding regions. For example, there were three inversions formed in the intron 5 of Pax7, a gene involved in developmental regulation, between 16.5 embryonic day and 12th month (Table S7c). The functional significance of these mutations remains to be determined.
Fig. 5: Examples of SV-disrupted genes.
a A deletion between Msh5 and 1700031A10Rik at the 4th month formed Msh5-1700031A10Rik out-of-frame fusion. b A duplication in Samd9 occurred at 16.5 embryonic days. c A t(7:12) translocation at the 4th month formed an out-of-frame Aldoa-Aldoart2 fusion. d Normal Rere structure VS the inversion at 16.5 embryonic days disrupted Rere structure.
KEGG pathway analysis revealed that the affected genes were enriched in multiple oncogenesis-related pathways, including estrogen signaling (Adcy2, Adcy3, Adcy6, Akt3, Atf2, Calm1, Ctsd, Gnai2, Gnas, Hsp90ab1, Itpr1, Kcnj5, Kcnj6, Krt10, Krt13, Krt20, Mmp2, Ncoa2, Pik3ca, Pik3cd, Plcb4, Sos1), cell cycle regulation (Anapc13, Ccnd2, Ccnd3, Cdc14b, Cdk6, Cdc27, Crebbp, Mcm7, Prkdc, Smad4, Smc1b, Stag1, Tfdp2, Ywhae, Zbtb17), cancer development (Akt3, Cdk6, Dvl2, Fgfr1, Fgf10, Mtor, Pik3ca, Pik3cd, Sos1), DNA damage repair (Brca1, Rad51, Rad51b, Rad51c, Sem1, Uimc1), Fanconi anemia (Rad51, Rad51c, Rev1, Rev3l), and base excision repair (Pole2, Pole3, Pole4) (Fig. S3, Table S7d). The abundant genes and pathways affected by the genome instability provided an environment in promoting cellular transformation towards cancer.
Certain genome instability in cancer cells originated at embryonic stageTaking advantage of the genome instability data available from the cancer developed in the same Brca1+/− mice [Fig. 1 in ref. 34], we compared the data between the non-cancer observed in our study and the cancer in Brca1-knockout mice. The results showed that the four major SV clusters in chromosome 4, 5, 11, and 13 observed in our study largely overlapped with those in the cancer cells, as exampled by the 11qD-qE cluster shared between the 10.5-day embryo and the cancer cells (Fig. 3). The overlaps indicated that these abnormalities in the cancer cells were likely originated earlier before the transformation of non-cancer cells into cancer cells.
The accumulated and de novo mutationsWhile the results above showed that the mutations were detected at the early embryonic stage, possibility may exist that the mutations detected could also include these accumulated from previous generations in the mutant strain considering that the mutant was generated more than two decades ago and propagated for many generations [17]. To test this possibility, we generated the Brca1 mutant and Brca1 normal mice by crossing the male and female mice of the same batch. We then sequenced the genomes at 10.5 and 16.5 embryonic days, and 1st month after birth. By using the sequences from kidney DNA, which is considered more stable than other tissue types, of the Brca1 mutant as the filter, we separated the accumulated mutations from the de novo mutation. We observed that around 2/3 of the mutations was the accumulated mutations (Table S8) and 1/3 were the de novo SVs, Indels, and CNVs, with SVs in particular, with similar patterns observed above as reflected by hotspot mutation formation, dynamic mutation change along developmental stages, breakpoints located at repetitive sequences (Table S9), affected genes including oncogenes, tumor suppressors, DNA damage repair genes, and immune function genes (Table S10), and mutated genes in non-homologous repair pathways (Table S11). The presence of de novo mutations after removing the accumulated mutations in the mutant mice confirmed that genome instability was indeed present at the embryonic stage in the heterozygotic Brca1 mutant genome.
Trp53 mutation played limited roles in Brca1 mutation-caused genome instabilityPrevious studies in the cancer developed in Brca1 mutant mice showed that Trp53 (TP53) mutation was required for the mutated Brca1 to cause genome instability [35, 36]. We introduced the Trp53+/− mutation to Brca1+/− to generate the Brca1+/− Trp53+/− mice. Using whole-genome sequencing, we collected the mutation data from Brca1+/− Trp53+/−, and compared the mutation data between Brca1+/− and Brca1+/− Trp53+/− mice. The results showed no significant differences for SVs and CNVs between the two groups but certain differences in indel (Table S12), indicating that Trp53 mutation was not essential for heterozygotic Brca1 mutation-caused genome instability in non-cancer cells.
留言 (0)