
記住我


Thalassemia is an inherited autosomal recessive blood disorder that is characteristic of abnormal hemoglobin production. According to the type of genetic defects, which result in reduced or absent synthesis of one or several globin peptide chains, thalassemia can be divided into α-, β-, and δβ-thal and hereditary persistence of fetal hemoglobin (HPFH), characterized by hemolytic anemia traits.1, 2 The human β-globin gene cluster, located on chromosome 11, is composed of five functional genes (HBE1, HBG2, HBG1, HBD, and HBB) with transcripts ε, Gγ, Aγ, δ, and β-globin.3 HPFH and δβ-thalassemia are relatively rare types of thalassemia caused by large deletions in the β-globin cluster involving δ- and β-globin genes, with or without the Aγ-globin gene, and are characterized by elevated fetal hemoglobin (HbF) in adults.4 Heterozygotes of HPFH are clinically asymptomatic, with a higher HbF, ranging from 15% to 30%. In contrast, δβ thalassemia heterozygotes show normal or mild values of hemoglobin (Hb) with a modest HbF ranging from 5% to 15%. However, when HPFH or δβ-thalassemia is coinherited with heterozygous β-thalassemia, patients may progress to a clinical phenotype of thalassemia intermedia or thalassemia major.5
To date, according to the Globin Gene Server homepage (http://globin.cse.psu.edu/hbvar/menu.html), more than 50 types of deletional HPFH and δβ-thalassemia have been reported in different ethnic groups and different regions. Molecular studies provide laboratory data to investigate the regulation of hemoglobin gene expression and search for the mechanism of Hb switching from fetal hemoglobin (HbF) to adult hemoglobin (HbA) and may play an important role in the pathogenesis of hemoglobin disorders during development.6 However, few reports have been reported on these mutations in Fujian population, which will aid in genetic counseling, as well as in prenatal diagnosis. Fujian Province, adjacent to Guangdong Province, located along the southeastern coastal regions of China, has a high prevalence of thalassemia.7 Therefore, the present large-scale study is designed to detect β-globin gene cluster deletions, characterize the genotypes, and analyze the phenotypes in Fujian Province, during the 14-year period from 2008 through 2021. Such a study may provide more data for genetic counseling and accurate prenatal diagnosis in this region and benefit for reducing the birth of babies with thalassemia major.
2 METHODS 2.1 Human subjectsA total of 55 001 subjects (18 268 men and 36 733 women) were screened for deletional HPFH/δβ-thalassemia using high-performance liquid chromatography (HPLC) from January 2008 to May 2016 and capillary electrophoresis from June 2016 to June 2021. Individuals showing HbF≥10.0%,8 before the blood transfusion, were suspected of carriers of deletional HPFH/δβ-thalassemia at the Outpatient Department of Fujian Maternity and Child Health Hospital during the period from January 2008 to June 2021 and were recruited for this study. The subjects, screened in this study, came from 9 cities across the province and had a mean age of 30 years (range from 2 to 66 years). This study was approved by the Ethics Review Committee of Fujian Maternity and Child Health Hospital. All participants wrote the informed consent following a detailed description of the purpose of the study. All subjects had no genetic relationship.
2.2 Screening for β-globin gene cluster deletionsApproximately 2 ml of the peripheral blood samples anticoagulated with EDTA-K2 from 55 001 subjects was used for analysis of blood cell parameters on a Sysmex XN-2000 automatic hematology analyzer (Sysmex). The hemoglobin components and levels of 34 128 subjects were performed using high-performance liquid chromatography (HPLC) with a VARIANT II TURBO Hemoglobin Testing System (Bio-Rad Laboratories, Inc.), and the remaining samples (including 20 873 subjects) were analyzed using an automated capillary electrophoresis system (CapillaryS 2, software version 6.2; Sebia).
2.3 Molecular analyses for β-globin gene cluster deletionsGenomic DNA was extracted from the peripheral blood samples using a genomic DNA isolation kit (Qiagen) following the manufacturer's instructions. Detection of the copy number variation in the β-globin gene cluster (NC_000011.10) was performed using the SALSA multiplex ligation-dependent probe amplification (MLPA) P102-B1 HBB assay (MRC-Holland), and thalassemia was definitively diagnosed using Gap-PCR with the deletional β-globin gene cluster detection kit (Shenzhen Yishengtang Biological Products Co., Ltd.). The detection results, including two common deletional HPFH, Chinese Gγ(Aγδβ)0 thalassemia and the Southeast Asia HPFH (SEA-HPFH) deletion,9 and the 1357 bp deletion (NG-000007.3:g.69997-71353 del 1357),10 were processed using MRC-Cofalyser version 9.4 (MRC-Holland), following the manufacturer's instructions. The three common deletional α-thalassemia (--SEA/αα, -α3.7/αα, and -α4.2/αα) were detected using Gap-PCR with the thalassemia gene detection kit (Shenzhen Yishengtang Biological Products Co., Ltd.),11 and detection of the point mutations in the three nondeletional α-thalassemia(αCSα/αα, αQSα/αα, and αWSα/αα) and the 17 common β-thalassemia was performed using reverse dot-blot hybridization (RDB) with the thalassemia gene detection kit (Shenzhen Yishengtang Biological Products Co., Ltd.) following the manufacturer's instructions.12
2.4 Statistical analysisThe hematological parameters of subjects with different genotypes were described using the mean and standard deviation. Associations between hematological and electrophoretic characterizations and the variant genotypes were assessed by a nonparametric Kruskal-Wallis test, presented as median (95% confidence interval). p values less than 0.05 were considered statistically significant. SPSS version 20.0 was used for statistical analysis.
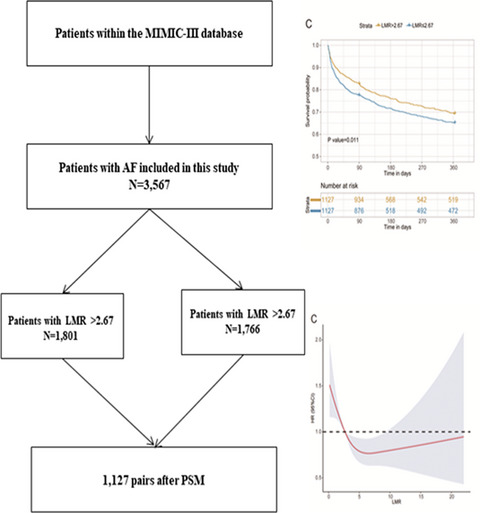
3 RESULTS 3.1 Prevalence of common β-globin gene cluster deletionsAmong 55 001 subjects, 142 cases with HbF (≥10%) were enrolled to characterize the molecular basis of β-globin gene cluster deletions in our study, including 47 men and 95 women aged 1 to 44 years. 22 cases were definitively diagnosed by MLPA (Figure 1) and Gap-PCR (Figure 2), with β-globin gene cluster deletions. The prevalence of common β-globin gene cluster deletions was 0.04% (22/55 001). Simultaneously, 62 cases with HbF (≥10%) were identified with common β-thalassemia, 29 cases were identified with common α-thalassemia, and 29 cases remained uncharacterized.

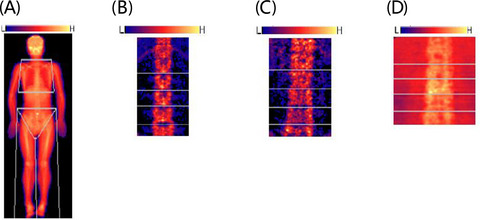
Screening for β-globin gene cluster deletions by MLPA, (A) SEA-HPFH deletion, (B) Chinese Gγ(Aγδβ)0-thal mutation, (C) 1357bp deletion(NG-000007.3:g.69997-71353 del 1357)

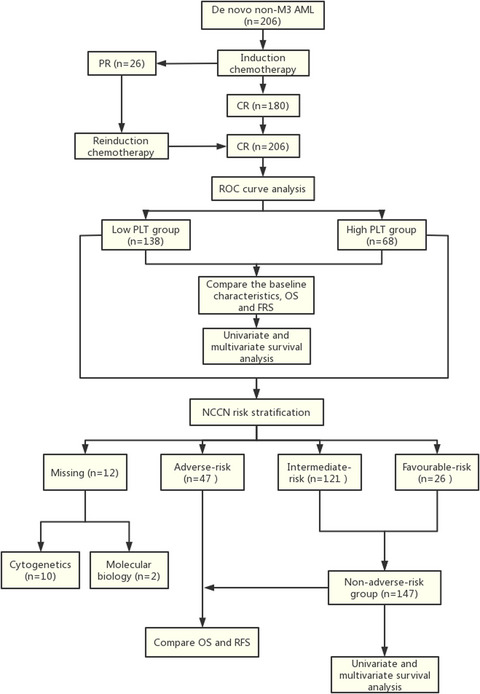
Diagnosis for β-globin gene cluster deletions by Gap-PCR, 1, Chinese Gγ(Aγδβ)0-thal mutation, 2, SEA-HPFH deletion, 3, 1357bp deletion (NG-000007.3:g.69997–71353 del 1357), 4, positive control (Chinese Gγ(Aγδβ)0-thal mutation), 5, negative control (The DNA template is from normal patients identified without β-globin gene cluster deletions), M, marker
3.2 Genotypes and phenotypes of β-globin gene cluster deletionsAs shown by MLPA (Figure 1) and Gap-PCR (Figure 2), three genotypes in 22 cases of β-globin gene cluster deletions were detected in our study. Among the 22 cases with β-globin gene cluster deletions, 10 cases were identified as heterozygous for Chinese Gγ(Aγδβ)0-thal mutations, 10 cases were heterozygous for SEA-HPFH, and one case was compound heterozygous for SEA-HPFH and α-thal mutations. Additionally, the 1357 bp deletion (NG-000007.3:g.69997-71353 del 1357) was detected in one case. The results of the hematological data of 22 cases are shown in Table 1, and a summary of the hematological data between different genotypes of β-globin gene cluster deletions (Kruskal-Wallis test) is presented in Table 2. Cases with heterozygous β-globin gene cluster deletions showed a range of phenotypes from normal to mild hypochromia and microcytosis. The levels of MCV, MCH, and HbA2 in male patients with heterozygous of Chinese Gγ(Aγδβ)0-thal were statistically lower than the male patients with SEA-HPFH deletion (p < 0.05), and the levels of HbA in male patients with heterozygous of Chinese Gγ(Aγδβ)0-thal were statistically higher than the male patients with SEA-HPFH deletion(p < 0.05). The levels of HbA2 in female patients with heterozygous of Chinese Gγ(Aγδβ)0-thal were statistically lower than the female patients with SEA-HPFH deletion (p < 0.05). The levels of MCV and MCH in female patients with heterozygous of Chinese Gγ(Aγδβ)0-thal were slightly lower than the female patients with SEA-HPFH deletion (p > 0.05), and the levels of HbA in female patients with heterozygous of Chinese Gγ(Aγδβ)0-thal were slightly higher than the female patients with SEA-HPFH deletion (p > 0.05). For patients who were compound heterozygous for SEA-HPFH deletion with α-thal mutation (ααWS/αα), the hematological and electrophoretic data were almost the same as data in patients with the SEA-HPFH deletion (p > 0.05). For patients with the 1357 bp deletion, the hematological and electrophoretic data were almost the same as other types (p > 0.05).
TABLE 1. Summary of the hematological and electrophoretic characterization of 22 cases with β-globin gene cluster deletions between different genotypes (Mean +/− SD) α-Genotype β-Genotype Gender No. Hb MCV MCH HbA HbA2 HbF αα/αα βN/βChinese Gγ(Aγδβ)0 F 5 116 ± 9.8 73.6 ± 3.9 25.1±1.3 79.1±2.8 2.5±0.2 18.4±2.9 M 5 143.0 ± 10.0 69.4 ± 4.5 22.9 ± 1.1 79.7 ± 2.4 2.6 ± 0.3 17.7 ± 2.6 F+M 10 129.8 ± 16.9 71.3 ± 4.6 23.9 ± 1.6 79.4 ± 2.5 2.5 ± 0.2 18.1 ± 2.7 αα/αα βN/βSEA−HPFH F 5 111.8 ± 12.0 77.2 ± 6.3 25.5 ± 1.8 73.8 ± 4.7 3.8 ± 0.5 22.3 ± 5.0 M 5 147.2 ± 9.5 77.9 ± 4.8 25.3 ± 0.5 75.8 ± 3.4 4.6 ± 0.4 19.6 ± 3.5 F+M 10 129.5 ± 21.3 77.5 ± 5.3 25.4 ± 1.3 74.8 ± 4.0 4.2 ± 0.6 21.0 ± 4.3 ααWS/αα βN/βSEA−HPFH F 1 119 75.9 23.8 74.2 4 21.8 αα/αα βN/β1357bp deletion F 1 116 72.6 25.2 80.9 6.3 13.6 Abbreviations: F, Female; M, Male. TABLE 2. Summary of the hematological and electrophoretic characterization of 22 cases with β-globin gene cluster deletions between different genotypes (Kruskal-Wallis test) α-Genotype β-Genotype Gender No. Hb (g/L) MCV (fl) MCH (pg) HbA (%) HbA2 (%) HbF (%) F 5 116.5.0 (100.8, 132.2) 73.6 (67.4, 79.9) 25.1 (22.9, 27.2) 79.2 (74.0, 84.4) 2.6 (2.3, 2.8) 18.2 (12.9, 23.6) αα/αα βN/βChinese Gγ(Aγδβ)0 M 5 143.0 (127.1, 158.9)a 71.1 (66.7, 75.5) 23.1 (21.3, 25.0) 79.4 (75.2, 83.7) 2.6 (2.1, 3.0) 18.0 (13.2, 22.7) F 5 111.8 (96.9, 126.6) 77.2 (69.4, 85.0) 25.5 (23.3, 27.8) 73.8 (68.0, 79.6) 3.8 (3.2, 4.4) 22.3 (16.1, 28.5) αα/αα βN/βSEA−HPFH M 5 147.2 (135.4, 159.0) 77.9 (72.0, 83.8) 25.3 (24.7, 25.9) 75.8 (71.6, 80.0) 4.6 (4.1, 5.2) 19.6 (15.2, 23.9) ααWS/αα βN/βSEA−HPFH F 1 119.0 75.9 23.8 74.2 4 21.8 αα/αα βN/β1357bp deletion F 1 116.0 72.6 25.2 80.9 6.3 13.6 p-valueb 0.461 0.028 0.009 0.027 0.009d 0.347 p-valuec 0.821 0.610 0.643 0.114 0.031d 0.183 4 DISCUSSIONAs the relatively rare types of thalassemia, HPFH and δβ-thalassemia, caused by large deletions in the β-globin cluster involving the δ- and β-globin genes, with or without the Aγ-globin gene, will affect the expression of the γ-globin genes.13 To date, more than 50 types of deletional HPFH and δβ-thalassemia have been reported in different ethnic groups and different regions.14 In our study, we found that in Fujian Province, the most common β-globin cluster deletions are the Chinese Gγ(Aγδβ)0, SEA-HPFH, and 1357 bp deletion. Here, we investigated the prevalence, molecular characterization, and phenotype of β-globin gene cluster deletions in Fujian individuals, which will be vital for genetic counseling and accurate prenatal diagnosis in this region to reduce the birth of babies with thalassemia major. To our knowledge, this is the first report on β-globin cluster deletions associated with hematological data in Fujian Province, a southeastern coastal region in China.
In this study, among 55 001 subjects, 142 patients with HbF (≥10%) were enrolled to characterize the molecular basis of β-globin gene cluster deletions, and 22 cases were definitively diagnosed with β-globin gene cluster deletions. The prevalence of common β-globin gene cluster deletions was 0.04% (22/55 001), which was different from other regions in China, slightly higher than the Yunnan region (0.024%),15 and lower than the Guangxi region (0.21%).9 All 22 patients in our study with heterozygous β-globin gene cluster deletions showed a range of phenotypes from normal to mild hypochromia and microcytosis. Some differences in hematological and electrophoretic data are shown in this study. The hemoglobin levels in cases with heterozygous for Chinese Gγ(Aγδβ)0-thal were slightly higher than in cases with SEA-HPFH deletion, whereas other red blood cell indices, such as MCV and MCH, were, in different degree, lower than in patients with the SEA-HPFH deletion across gender, which was slightly different from previous reported.16 This might be because the cases in this study were limited and the previous report did not analyze across gender. Moreover, the hemoglobin A2 levels in patients who were heterozygous for Chinese Gγ(Aγδβ)0-thal were statistically lower than in cases with SEA-HPFH deletion(p < 0.05). For patients who were compound heterozygous for SEA-HPFH deletion with α-thal mutation (ααWS/αα), the hematological and electrophoretic data were almost the same as in patients with the SEA-HPFH deletion (p > 0.05). For patients with the 1357 bp deletion, the hematological and electrophoretic data were almost the same as other types (p > 0.05).
In our study, among the 142 patients with HbF (≥10%), 62 patients were identified with common β-thalassemia. In β-thalassemia, where β-globin is deficient, increased γ-globin expression reduces the imbalance of the α- and β-globin chains, which underlies the pathophysiology of anemia in this condition. Moreover, KLF1 mutations were selectively reported in the presence of β-thalassemia to increase the production of HbF.17 Patients with β-thalassemia showed an increased level of HbF, which may be the result of the polymorphism of Xmnl and p. Ser102Pro (c.304T> C) in the Krüppel-like factor (1 KLF1) gene, or because of the number of α-gene deletions.18-21 In the 29 cases with common α-thalassemia, 26 cases were confirmed with -α4.2/αα and screened by capillary electrophoresis, which was suspected to be compound with HbQ-Thailand and confirmed by DNA sequencing of the α genes (data not shown).22, 23 The uncharacterized cases may be explained by a nondeletional point mutation in the γ-globin genes and regulatory factors such as KLF1, B cell CLL/lymphoma 11A (BCL11A), zinc-finger and BTB-domain-containing 7A (ZBTB7A), and GATA1.24-28
In conclusion, in Fujian Province, the prevalence of common β-globin gene cluster deletions was 0.04%. What's more, the most common β-globin cluster deletions are the Chinese Gγ(Aγδβ)0 and SEA-HPFH.
ACKNOWLEDGEMENTWe thank all patients for their cooperation.
CONFLICT OF INTERESTThe authors confirm that they have no competing interests.
AUTHOR CONTRIBUTIONSMeihuan Chen, Liangpu Xu, and Hailong Huang designed the study and prepared the study. Min Zhang, Lingji Chen, Na Lin, and Yan Wang collected the literature, collected the data, and prepared the study. All authors approved the final study.
留言 (0)