
記住我
Medicinal chemistry is the interdisciplinary field that focuses on the design, development, and optimization of pharmaceutical compounds. It combines principles from chemistry, biology, and pharmacology to create bioactive molecules that can effectively treat diseases (Francesco, 2023). Key research contents include the synthesis of new compounds, structure-activity relationship (SAR) studies, optimization of drug properties (such as solubility and bioavailability), and the evaluation of pharmacological effects. The discovery and design of small molecule drugs have been pivotal in the field of medicinal chemistry, serving as the cornerstone for developing new therapeutics to combat a myriad of diseases (Campbell et al., 2018). Small molecules have historically played a critical role in the development of effective treatments for a wide range of diseases, including cancer, infectious diseases, and chronic illnesses (Zhong et al., 2021; Davis et al., 2020; Parra Sánchez et al., 2022). Despite their significance, the process of small molecule drug discovery is fraught with challenges, such as high attrition rates in clinical trials, the complexity of biological systems, and the need for precision-targeted therapies (Zhong et al., 2021; Keserü and Makara, 2009; Bedard et al., 2020). As such, there is a pressing need for innovative approaches that can enhance the efficiency and success of small molecule drug design.
One of the primary challenges in the drug discovery process is the vast chemical space that needs to be navigated (Saldívar-González and Medina-Franco, 2022). Traditional methods often rely on trial-and-error approaches, which are not only time-consuming but also resource-intensive. Additionally, the complexity of biological systems and the diverse nature of diseases necessitate a more targeted and strategic approach to drug design. In this context, several new methodologies have emerged, promising to transform the landscape of small molecule drug discovery.
Click Chemistry has garnered significant attention as a powerful tool in drug discovery. It enables the rapid synthesis of diverse compound libraries through highly efficient and selective reactions (Zhao et al., 2024; Wilson Lucas et al., 2023). Click chemistry’s modular nature allows for the straightforward incorporation of various functional groups, facilitating the optimization of lead compounds and enabling the creation of complex structures from simple precursors (Jiang et al., 2019). This approach not only accelerates the discovery process but also enhances the likelihood of identifying compounds with desirable pharmacological properties.
Targeted Protein Degradation (TPD) strategies represent another groundbreaking advancement in small molecule drug design (Wells and Kumru, 2024). Unlike traditional inhibitors that aim to block protein activity, TPD technologies employ small molecules to tag undruggable proteins for degradation via the ubiquitin-proteasome system or autophagic-lysosomal system (Song et al., 2023). This novel approach provides a means to address undruggable targets and offers a new therapeutic paradigm for conditions where conventional small molecules have fallen short. By harnessing the body’s own degradation machinery, TPD strategies enable the selective removal of disease-associated proteins, thereby providing an innovative pathway for drug discovery.
DNA-Encoded Libraries (DELs) have emerged as a widely used technology that allows for the high-throughput screening of vast chemical libraries (Peterson and Liu, 2023). DELs utilize DNA as a unique identifier for each compound, facilitating the simultaneous testing of millions of small molecules against biological targets (Dockerill and Winssinger, 2023). This technology not only streamlines the identification of potential drug candidates but also allows for the exploration of chemical diversity in an unprecedented manner. The ability to rapidly probe the interaction of small molecules with target proteins enhances the efficiency of lead discovery and optimization, making DELs an essential tool in modern drug development.
Computer-Aided Drug Design (CADD) has also significantly influenced the drug discovery landscape by employing computational methods to predict the binding affinity of small molecules and specific targets (Sadybekov and Katritch, 2023; Jiménez-Luna et al., 2021) This approach significantly reduces the time and resources required for experimental screening (Giordano et al., 2022). With advancements in artificial intelligence (AI), CADD is becoming increasingly sophisticated, enabling researchers to simulate complex biological interactions and refine drug design more effectively (You et al., 2022). This predictive capability not only accelerates the discovery process but also enhances the precision of drug design, addressing the critical need for tailored therapies.
Each approach brings unique advantages, addressing specific challenges in the drug development pipeline and paving the way for innovative therapeutic solutions. The integration of these technologies promises to enhance the efficiency of drug discovery, reduce attrition rates in clinical trials, and ultimately lead to the development of more effective and targeted therapies. This review aims to provide a comprehensive overview of recent advancements in these approaches, to explore their underlying principles, and their potential impact on the future of medicinal chemistry. To illustrate the utility of these techniques, we highlight exemplary small molecules developed over the past decade, emphasizing their role in accelerating drug development. Furthermore, we summarize representative marketed drugs and investigational new drugs, illustrating these technologies' direct contributions to clinical practice. Finally, we discuss the future prospects and challenges of each approach. By fostering an understanding of these emerging strategies, we aim to inspire new research directions and collaborations that will further advance the field of medicinal chemistry.
2 Click chemistryClick chemistry, a combinatorial methodology first introduced by Professor Sharpless in 2001, revolutionized the rapid synthesis of C-X-C atom frameworks (Kolb et al., 2001). This approach has since become an essential tool in the medicinal chemist’s toolbox, offering significant advantages such as broad substrate scope, high yield, stereospecificity, operational simplicity, and the formation of only benign by-products that can often be removed without chromatography (Caselli et al., 2015; Le Droumaguet et al., 2022). Additionally, click reactions are conducted using environmentally benign solvents, further enhancing their utility in drug discovery (Shankaraiah et al., 2019). In contrast to traditional drug design, click chemistry employs modular reactions to efficiently create new drug-like molecules. Moreover, these reactions serve as versatile linkers for small molecules and proteins. An example is their application in the synthesis of proteolysis targeting chimeras (PROTACs), where click chemistry links two pharmacophores via a specific scaffold to produce compounds with high target affinity (Wurz et al., 2017). Click chemistry is also instrumental in constructing diverse compound libraries, offering a robust platform for screening bioactive molecules and potential therapeutic agents. Importantly, target-templated in situ click chemistry allows for the direct generation of hits within the binding pocket of a target, streamlining the discovery of enzyme inhibitors and other bioactive compounds. This powerful method significantly accelerates both the synthesis and screening processes in drug discovery (Kugler et al., 2023; Glassford et al., 2016).
2.1 The conventional click chemistryConventional click chemistry has found widespread application in various domains of drug discovery, facilitating the modular synthesis of new drug-like molecules, serving as a linker for PROTACs or pharmacophore groups, and enabling the rapid construction of compound libraries. This efficient, chemo-selective synthesis method allows for the coupling of molecular fragments under mild reaction conditions, making it highly desirable in medicinal chemistry. Key reactions within click chemistry include the Cu-catalyzed azide-alkyne cycloaddition (CuAAC), strain-promoted azide-alkyne cycloaddition (SPAAC), thiol-ene reactions, inverse electron demand Diels–Alder reactions (IEDDA), hydrazone click chemistry, and the recently emerging sulfur fluoride exchange (SuFEx) reaction (Shankaraiah et al., 2019; Kim and Koo, 2019; Barrow et al., 2019). Since its introduction in 2001, click chemistry has become a prominent research focus, driving innovation in the development of bioactive compounds and therapeutic agents (Cai et al., 2023).
2.1.1 Cu-catalyzed azide-alkyne cycloaddition (CuAAC)The discovery of Cu(I) catalyzed CuAAC in 2001 represented a pivotal advancement, transforming click chemistry from a theoretical concept into a widely accepted and practical methodology (Kolb et al., 2001). Among all click reactions, CuAAC has emerged as the most popular synthetic and coupling tool. This reaction offers exceptional application potential due to its ability to facilitate the functionalization of organic scaffolds with azides and alkynes (Jiang et al., 2019), which remain stable during subsequent transformations in the presence of highly functionalized biomolecules, molecular oxygen, water, and other common reaction conditions. In contrast to the uncatalyzed cycloaddition of azides and alkynes, which yields a mixture of 1,4- and 1,5-triazole regioisomers at elevated temperatures, CuAAC selectively combines organic azides and terminal alkynes to produce 1,4-disubstituted 1,2,3-triazoles exclusively under mild conditions (Figure 1A), thus eliminating the need to separate the 1,4- and 1,5-linked regioisomers using classical chromatographic techniques. The detailed reaction mechanisms underlying this transformation are illustrated in Figure 2A. Notably, the CuAAC reaction offers key advantages, including exceptional stereoselectivity, rapid reaction rates, and the ability to form intermolecular connections efficiently under mild conditions. These attributes have positioned CuAAC as a powerful and widely adopted tool in drug discovery. Compounds containing the 1,2,3-triazole moiety have demonstrated compelling biological activities, as illustrated by several approved drugs and drug candidates depicted in Figure 3A.
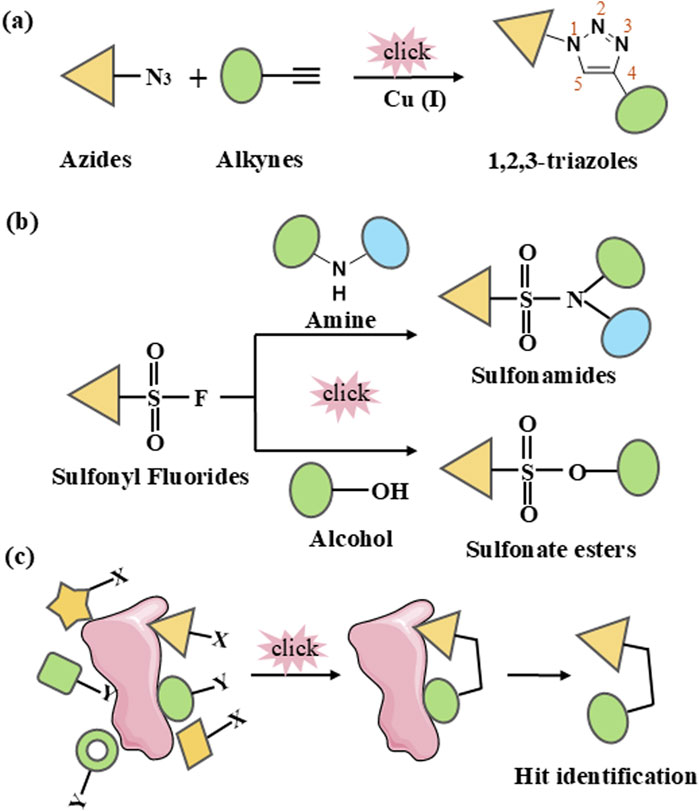
Figure 1. Schematic representation of click chemistry. (A) Cu-catalyzed azide-alkyne cycloaddition (CuAAC). (B) Sulfur (VI)-Fluoride Exchange (SuFEx). (C) In situ click chemistry approach.
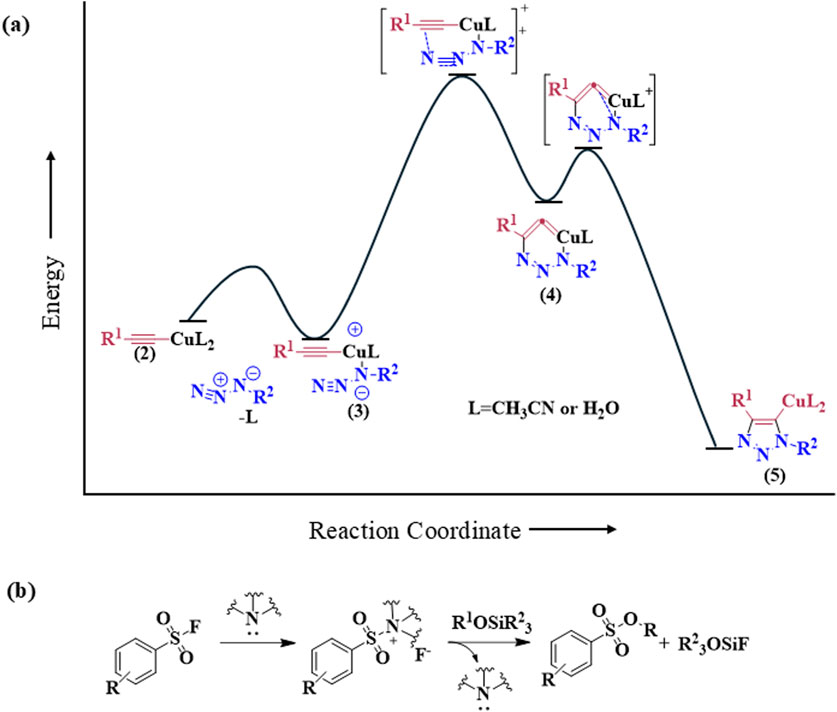
Figure 2. Reaction mechanisms of click chemistry of (A) Cu-catalyzed azide-alkyne cycloaddition (CuAAC) and (B) Sulfur (VI)-Fluoride Exchange.
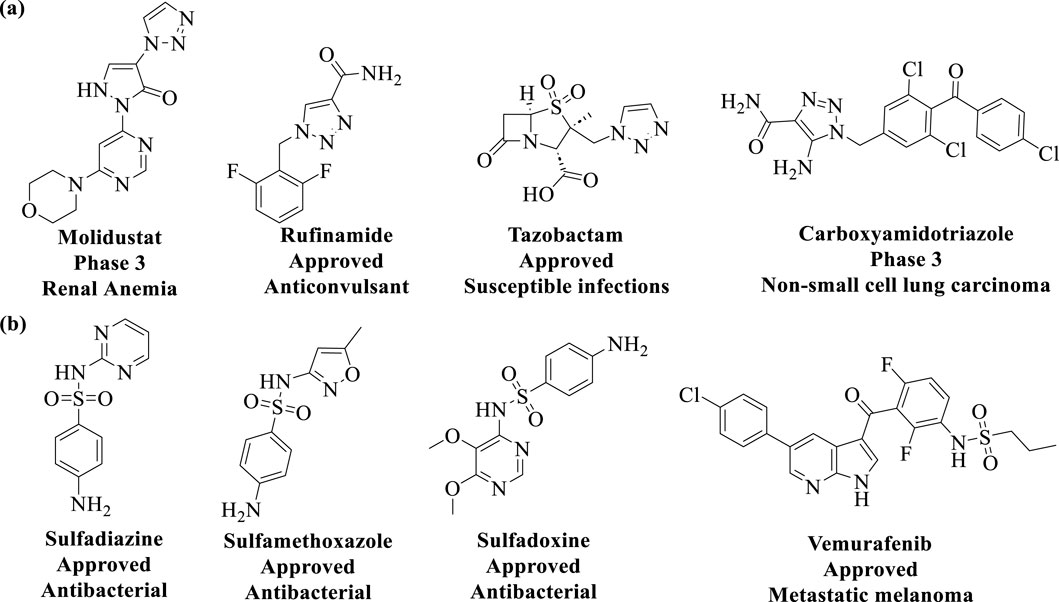
Figure 3. Chemical structures of approved drugs and drug candidates containing the (A) 1,2,3-triazole moiety, and (B) sulfonamides moiety.
Recent studies illustrate the utility of CuAAC click chemistry in drug discovery. For instance, Synta66 has been employed as a chemical probe to elucidate the role of store-operated calcium entry (SOCE) in cellular mechanisms. To identify drug-like SOCE inhibitors with improved pharmacokinetic profiles, Pirali et al. replaced the amide group in Synta66 with a triazole ring. Among the synthesized 1,2,3-triazoles, compound #1 (Figure 4A) exhibited significantly higher aqueous solubility (1,528 μg/mL vs. 0.28 μg/mL) compared to Synta66. Furthermore, compound #1 demonstrated potent inhibitory activity at nanomolar concentrations, favorable pharmacokinetic properties, and in vivo efficacy in a mouse model of acute pancreatitis (Serafini et al., 2020). In another study, Manera et al. designed, synthesized, and functionally characterized the first bitopic ligands for the cannabinoid receptor CB2 (CB2R). These ligands were synthesized through a click chemistry reaction between azido and alkyne derivatives. The most promising bitopic ligand, FD-22a (Figure 4A), exhibited anti-inflammatory activity in a human microglial cell inflammatory model and demonstrated antinociceptive effects in vivo in a mouse model of neuropathic pain (Gado et al., 2022). Additionally, Cee et al. reported a click chemistry approach as a reliable linking strategy for the synthesis of a 10-membered library of PROTACs (Figure 4A). This method enables the parallel synthesis of PROTAC libraries, contingent upon the availability of the requisite azides and alkynes. Conceptually, this platform represents a powerful new tool for accessing diverse PROTAC libraries, with principles that can be readily applied to other ligases and target proteins (Wurz et al., 2017).
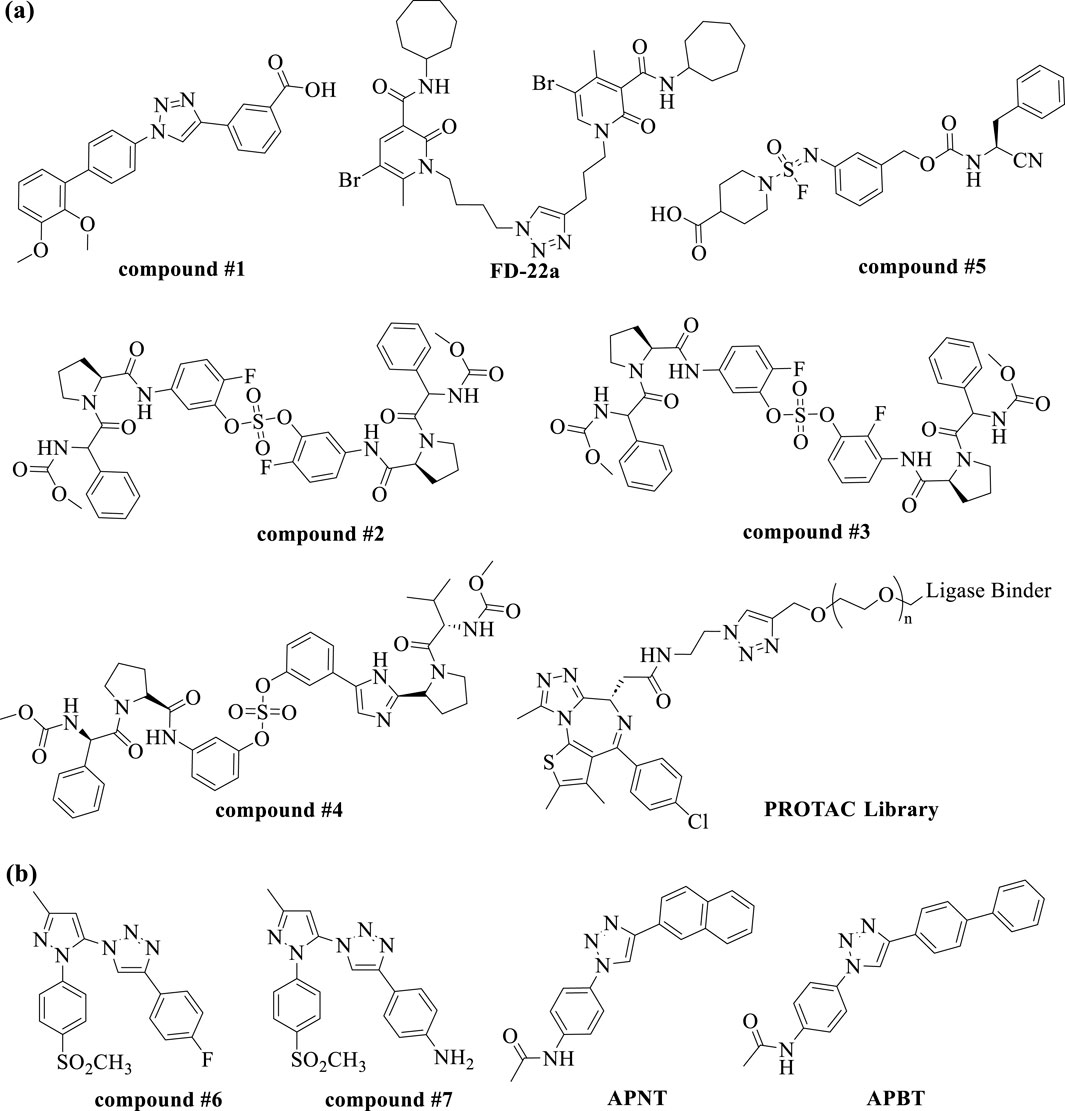
Figure 4. Chemical structures of small molecules discovered from (A) the conventional click chemistry and (B) in situ click chemistry.
2.1.2 Sulfur (VI)-fluoride exchange (SuFEx)In 2014, Sharpless and colleagues introduced the next advancement in click chemistry: SuFEx (Click-II) (Figure 1B). The detailed reaction mechanisms are shown in Figure 2B. SuFEx represents a recent set of ideal click chemistry transformations, characterized by metal-free reaction conditions. This approach utilizes sulfuryl fluoride (SO2F2) and thionyl tetrafluoride (SOF4) to synthesize two key S–F motifs: arylfluorosulfates (Ar–O–SO2–F) and iminosulfur oxydifluorides (R–NSOF2) (Barrow et al., 2019). Although the potential of SuFEx in drug discovery is just beginning to be explored, it holds promise. In contrast to the CuAAC reaction, which has been employed in proof-of-concept studies for lead optimization, only a limited number of drugs feature the 1,2,3-triazole linkage. Sulfonamides, however, are a common structural motif in drug design and represent the primary sulfur-based feature in clinically approved medications (Figure 3B). More than 150 sulfonamide drugs are available on the market (Zhao et al., 2019), encompassing a range of therapeutic activities including antibacterial, antitumor, anti-obesity, anti-thyroid, and analgesic effects for neuropathic pain. Furthermore, sulfonate esters (R–SO2–OR) and sulfate diesters (R–OSO2–OR) serve as excellent bioisosteric substitutes for carboxylic acids and esters. Despite their greater lipophilicity, these sulfonyl compounds maintain a polarized S–O bond, facilitating robust electrostatic interactions with target proteins (Hong et al., 2019). This positions SuFEx as a promising tool for the modular synthesis of functional libraries in drug discovery.
SuFEx click chemistry has emerged as a valuable tool in drug discovery. Kim, Jang, and colleagues successfully utilized SuFEx reactions in a click chemistry approach to synthesize biaryl sulfate core derivatives, demonstrating their efficacy as potent inhibitors of hepatitis C virus (HCV) nonstructural protein 5A (NS5A). Among the synthesized inhibitors, compounds #2, #3, and #4 (Figure 4A) exhibited impressive two-digit picomolar EC50 values against HCV genotype-1b (GT-1b) and single-digit or sub-nanomolar activities against the HCV genotype-2a (GT-2a) strain (You et al., 2018). A pioneering study by Wolan et al. showcased biocompatible SuFEx click chemistry as a proof-of-concept for a high-throughput process aimed at generating drug-like, biologically active molecules. Starting from a modest high-throughput screening hit against the bacterial cysteine protease SpeB, a SuFExable iminosulfur oxydifluoride [RN = S(O)F2] motif was introduced, leading to the rapid diversification into 460 analogs in overnight reactions. Direct screening of these products yielded drug-like inhibitors with up to 300-fold increased potency. Compound #5 (Figure 4A) is the first potent and selective SpeB inhibitor, useful for studying the protease’s role in cellular and animal models (Kitamura et al., 2020). More recently, the research groups of Moses et al. demonstrated the versatility and practicality of Accelerated SuFEx Click Chemistry (ASCC) for the late-stage derivatization of bioactive molecules. They employed ASCC to synthesize discrete compound libraries in a 96-well plate format, ensuring experimental consistency and significantly enhancing the efficiency of identifying potent hit molecules. Additionally, ASCC was utilized to create arrays of sulfonate ester-linked, high-potency microtubule-targeting agents (MTAs) that exhibited nanomolar anticancer activity against multidrug-resistant cancer cell lines. These findings highlight ASCC’s promise as a robust platform for advancing drug discovery (Homer et al., 2024).
2.2 In situ click chemistryThe discovery of bioactive compounds hinges on the iterative generation of SAR data derived from the preparation and biological assays of hit congeners. However, traditional methods are often time-consuming and labor-intensive, leading to slow and costly hit-to-lead transitions. Target-guided synthesis (TGS) is a synthetic strategy for enzyme inhibitors that involves the selective assembly of complementary functional groups based on biological targets (e.g., enzymes) serving as templates (Hu and Manetsch, 2010a). The application of click chemistry in kinetic TGS, known as in situ click chemistry, represents an innovative synthesis process where a biological target facilitates the assembly of its own inhibitor through the guided selection of suitable building blocks (Hu and Manetsch, 2010a; Bhardwaj et al., 2017) (Figure 1C). This approach not only results in compounds with high binding affinity for the target but also offers a powerful strategy for rapidly preparing and characterizing compound libraries of bioactive molecules, greatly improving the efficiency of drug discovery. Fragment-based drug design plays a crucial role in drug discovery. In situ click chemistry provides a unique strategy for fragment modification and optimization. In this approach, chemical ligation depends on the close proximity and optimal spatial arrangement of reactive fragments (Bhardwaj et al., 2017). When these fragments bind simultaneously to the target’s binding sites, they facilitate the formation of irreversible bonds. Consequently, the selection of suitable combinatorial fragment libraries is a critical factor for the success of this method.
The general protocol for assembling and screening focused combinatorial fragment libraries using in situ click chemistry involves four key steps. First, suitable fragments are selected, typically featuring a common warhead—a core structure for interaction with a specific class of biological targets. These fragments should exhibit a variety of substituents and functional groups, as greater skeletal and stereochemical diversity within the chemical space increases the likelihood of identifying effective hits. Additionally, the fragments need to possess desirable properties such as stability, solubility, and cellular permeability to ensure compatibility with biological systems (Wang et al., 2016; Rzuczek et al., 2014; Hu and Manetsch, 2010b). Second, the reactant fragments are synthesized to include reactive groups such as azides, alkynes, or sulfonyl fluorides, which serve as warheads in the click chemistry reactions. Third, in situ screening is conducted, typically in 96- or 384-well plates, enabling high-throughput screening without further purification, thus streamlining the drug discovery process. Finally, interactions between the fragment molecules and target compounds are assessed based on preliminary screening data, such as percentage inhibition, with promising hits further validated through IC50 determination to confirm their potency (Wang et al., 2016).
In recent years, the rapid assembly and in situ screening of focused combinatorial fragment libraries using CuAAC and SuFEx click chemistry have emerged as robust and efficient strategies for generating bioactive molecules. In 2015, Wuest and colleagues demonstrated for the first time the use of CuAAC click chemistry in conjunction with the cyclooxygenase-2 (COX-2) active site as a reaction vessel for the in situ generation of highly specific inhibitors. This study led to the discovery of two highly potent and selective COX-2 isozyme inhibitors (compounds #6 and #7) (Figure 4B), which exhibited significantly greater in vivo anti-inflammatory activity compared to widely used selective COX-2 inhibitors. These findings provide a valuable tool for the economical and rapid screening of potential drug candidates (Bhardwaj et al., 2017). In 2016, Wang and colleagues introduced two cell-permeable O-GlcNAc transferase (OGT) inhibitors, APNT and APBT (Figure 4B), developed from low-activity precursors (IC50 > 1 mM) through a strategy termed “tethering in situ click chemistry” (TISCC). Among these, APNT exhibited a significantly enhanced inhibitory activity, with an IC50 of 66.7 ± 0.8 μM, representing over a 60-fold improvement. Notably, APNT’s potency was comparable to that of benzoxazolinone, an irreversible OGT inhibitor. However, benzoxazolinone’s high reactivity made it unsuitable as a selective OGT inhibitor for cellular applications. In contrast, both APNT and APBT effectively suppressed O-GlcNAcylation in cells without causing significant cytotoxicity. This highlights TISCC as a promising approach for optimizing low-activity precursors with millimolar IC50 values into potent and selective inhibitors (Wang Y. et al., 2017).
2.3 Challenges of click chemistry in drug discoveryClick chemistry offers several advantages, including insensitive to oxygen and water, high yields, regioselectivity, and stereoselectivity. These properties enable researchers to rapidly prepare target compounds and build compound libraries. Furthermore, in situ click chemistry leverages enzymes as reaction templates, taking advantage of their favorable conformations under physiologically relevant conditions. This approach selectively connects individual building blocks to synthesize novel enzyme inhibitors. However, the application of click chemistry in drug discovery also presents challenges and should not be regarded as a universal solution. Firstly, when generating large numbers of compounds at a rapid pace, it remains uncertain whether click chemistry consistently yields compounds with desirable drug-like properties. As a result, further optimization of biological activity and drug-like characteristics can also be time-consuming. While copper species are among the simplest and most useful catalysts for the CuAAC click reaction, their introduction into biological systems raises concerns regarding potential toxicity, which may interfere with the screening of compound libraries utilizing biological targets as templates. In contrast, the SuFEx reaction is a metal-free procedure that shows promise for library screening. The synthesis of 1,2,3-triazole rings as pharmacophores or bioisosteres via CuAAC has significant potential in drug design for various diseases. However, the 1,2,3-triazole ring itself is not commonly utilized as a pharmacophore and is rarely found in marketed drugs, indicating certain limitations in its application as a drug molecule. Conversely, sulfonamides are a prevalent feature in drug structures and serve as the primary sulfur-based motif in clinically approved drugs, with over 150 sulfonamide drugs available in the market. Despite their stability, ease of synthesis, and potential as pharmacophores, sulfur-ester linkages remain notably underrepresented in drug molecules. Lastly, despite the decades since its inception, click chemistry encompasses only a limited number of reactions, resulting in the identification of multiple hits and leads for specific targets, but relatively few marketed drugs have been successfully discovered through this approach. Consequently, while click chemistry presents both advantages and disadvantages in medicinal chemistry and drug discovery, it also faces numerous challenges. Ongoing developments hold the promise of advancing this synthetic technique to meet the preclinical and clinical requirements of various systemic and localized diseases.
3 Targeted protein degradation (TPD) strategiesTargeting pathogenic proteins with small molecule inhibitors has become a widely adopted strategy for treating various diseases. However, many intracellular proteins are deemed “undruggable” due to the absence of accessible active sites, posing a significant challenge in the design and development of effective small molecule inhibitors (Xie et al., 2023). TPD is an emerging concept in drug discovery, first introduced in 1999 (Wickner et al., 1999). Recently, TPD has gained traction as a novel approach for targeting these previously undruggable proteins, utilizing proteasomal and lysosomal pathways (Ding et al., 2020; Zhang C. et al., 2024). Unlike traditional inhibitors, small molecule degraders do not require continuous exposure of the protein’s binding site, which enhances their therapeutic potential. Additionally, many degraders, such as PROTACs, operate catalytically, allowing for lower dosages to achieve the desired effects (Bondeson et al., 2015). Most TPD strategies, including PROTACs and molecular glues, primarily rely on the ubiquitin–proteasome system (UPS) and predominantly target intracellular proteins. In contrast, lysosome-dependent TPD strategies can degrade membrane proteins, extracellular proteins, and protein aggregates, significantly broadening the range of substrates that can be targeted (Zhao et al., 2022). The proper functioning of these two pathways is crucial for preventing or treating a variety of diseases, including tumors, neurodegenerative disorders, autoimmune diseases, and metabolic syndrome. Therefore, employing proteasomes and lysosomes to target pathogenic proteins represents a promising strategy in addressing these complex health challenges.
3.1 Targeted protein degradation via proteasomeThe UPS is a crucial pathway for protein degradation, playing a vital role in maintaining cellular homeostasis by facilitating the degradation of over 80% cellular proteins (Chen et al., 2016). This degradation process targets damaged proteins or those that are no longer needed. Ubiquitination is achieved through a cascade involving three key enzymes: an E1 activating enzyme, an E2 conjugating enzyme, and an E3 ligase. The E1 enzyme binds to a ubiquitin molecule in an ATP-dependent manner and subsequently transfers it to the E2 enzyme. The E3 ligase then catalyzes the transfer of the ubiquitin from E2 to the target substrate, leading to the polyubiquitination of the substrate (Eldridge and O'Brien, 2009). The accumulation of the polyubiquitin chain on a targeted protein serves as a signal for degradation by the proteasome. PROTACs and molecular glues are two prominent technologies that leverage the UPS for the targeted degradation of specific proteins of interest (POI), and will be the focus of our discussion.
3.1.1 Proteolysis-targeting chimeras (PROTACs)The UPS, exemplified by PROTACs, offers a powerful strategy for degrading otherwise undruggable POI. PROTACs are synthetic heterobifunctional molecules composed of an E3-recruiting ligand, a POI-targeting warhead, and a flexible linker that connects the two (Konstantinidou et al., 2019). By promoting the formation of a ternary complex comprising the POI, PROTAC, and E3 ligase, these compounds enhance the ubiquitination of the POI, leading to its degradation via the UPS (Figure 5A). Unlike traditional inhibitors, PROTACs operate through an event-driven mechanism, acting as catalysts for selective protein degradation—one molecule of PROTAC can induce the ubiquitination of multiple target protein molecules (Burslem and Crews, 2020). Initially demonstrated by Crews and colleagues in 2001, the concept of PROTACs has since evolved into various innovative degradation technologies targeting kinases, nuclear receptors, epigenetic proteins, misfolded proteins, and even RNAs (Sakamoto et al., 2001; Schreiber, 1992). This broadens the spectrum of potential targets and enhances clinical applications for treating cancer, neurodegenerative diseases, and viral infections. Consequently, PROTAC technology is advancing into clinical settings, with over 15 targeted degraders currently in clinical trials (Figure 6A).
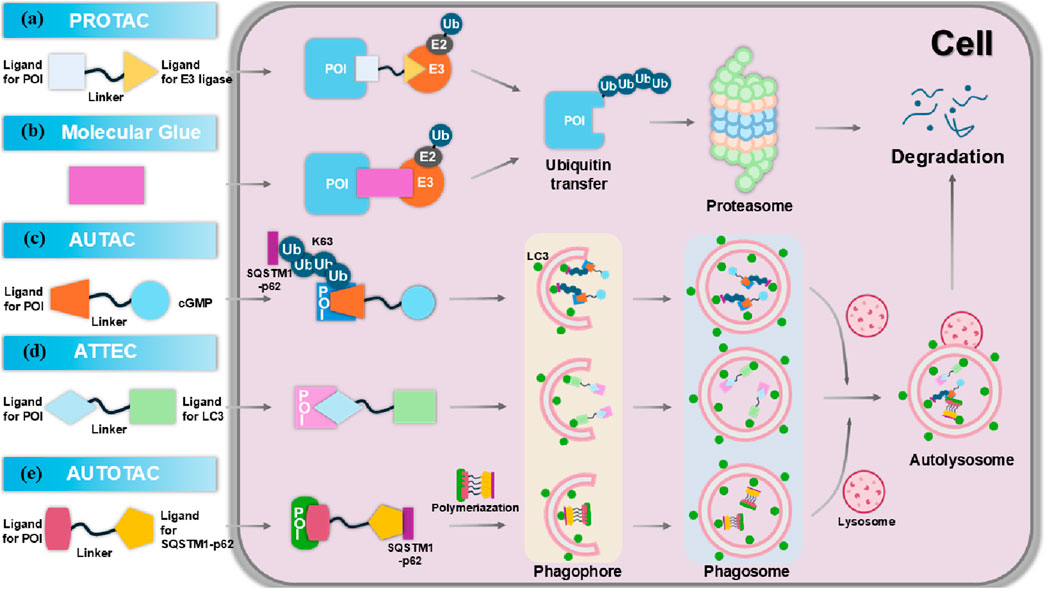
Figure 5. The molecular mechanism of targeted protein degradation (TPD). (A) Proteolysis targeting chimeras (PROTACs) are composed of an E3 ligase-targeting ligand, a linker, and a proteins of interest (POI)-binding ligand. By simultaneously interacting with both the POI and the E3 ligase, PROTACs promote the polyubiquitination of the POI, marking it for degradation via the proteasome. (B) Molecular glues bind to the E3 ubiquitin ligase, or the POI, facilitating their interaction and triggering the ubiquitination and degradation of the POI. (C) Autophagy-targeting chimeras (AUTACs) consist of a POI-targeting warhead, a linker, and a cGMP-based degradation tag. This tag promotes K63 polyubiquitination of the POI, leading to selective degradation via autophagy in the lysosome. (D) Autophagosome-tethering compounds (ATTECs) bind LC3 and the POI, (E) while AUTOphagy-TArgeting Chimeras (AUTOTACs) bind p62 and the POI, driving autophagosome formation, which then merge with lysosomes to degrade the targeted POI.

Figure 6. Chemical structures of proteasome-based degraders. Representational PROTACs currently in (A) clinical trials and (B) preclinical trials. Representational molecular glues currently in (C) clinical trials and (D) preclinical trials. Degradation tag and proteins of interest (POI) warheads are respectively marked in blue and red.
The PROTAC technology represents a promising therapeutic modality for treating various diseases. Here, we highlight some PROTACs that target specific proteins for degradation. A significant achievement in PROTAC technology is the development of orally bioavailable PROTACs that have progressed to clinical trials. Notably, the androgen receptor degrader ARV-110 (Clinical Trial No. NCT03888612) and the estrogen receptor degrader ARV-471 (Clinical Trial No. NCT05654623) have entered phase II and III clinical trials for prostate and breast cancer, respectively. The PROTAC approach, widely exploited in cancer research, also holds promise for antiviral development. In 2022, the Marazzi and Jin groups identified FM-74-103 (Figure 6B), a small-molecule degrader that inhibits infections from Influenza A virus (IAV), syndrome coronavirus 2 (SARS-CoV-2), and cytomegalovirus (CMV). FM-74-103 selectively degrades human G1 to S phase transition 1 (GSPT1), a translation termination factor, showcasing its potential as a host-directed antiviral that can degrade key factors controlling both RNA and DNA virus replication in host cells. This work highlights the broad utility of the PROTAC platform for rational design and development of next-generation antivirals (Zhao et al., 2023). Moreover, PROTACs can target undruggable proteins, significantly expanding the therapeutic prospects for refractory diseases. Tau protein accumulation is a hallmark of Alzheimer’s disease (AD) and related tauopathies; however, tau is a natively unfolded protein lacking well-defined folds and active sites. In 2021, Wang et al. discovered C004019 (Figure 6B), a novel small-molecule PROTAC that selectively promotes tau clearance, significantly improving synaptic and cognitive functions in various hTau cell models and in AD-like hTau and 3xTg transgenic mice. This work demonstrates that PROTACs can effectively induce the degradation of undruggable proteins, offering promising therapeutic avenues for AD and related tauopathies (Wang W. et al., 2021).
3.1.2 Molecular glueThe concept of molecular glue was first introduced Harvard chemical biologist Stuart Schreiber in the early 1990s (Schreiber, 1992). As a linker-free scaffold, molecular glues induce, stabilize, or enhance interactions between E3 ligases and their POI by modifying the surface of the E3 ubiquitin ligase, thereby promoting the ubiquitination of the POI (Zhou et al., 2023) (Figure 5B). This modality effectively hijacks the ubiquitin-proteasome pathway to degrade traditionally challenging therapeutic targets, thereby broadening the spectrum of druggable proteins. Unlike PROTACs, molecular glues are generally smaller (typically<500 Da) and lack clearly separable components like linkers or distinct chemical moieties for binding to each protein. While their smaller size and successful precedents suggest that molecular glues may exhibit better drug-like properties, their design poses significant challenges. However, the few molecular glues that have made it into clinical trials were largely discovered serendipitously (Ding et al., 2022) (Figure 6C). In 2014, Krönke et al. reported the first molecular glue, lenalidomide (a thalidomide analog), which induces the degradation of IKZF1/3 via the CUL4/CRBN pathway (Krönke et al., 2014). Since the discovery of thalidomide, over ten small molecular glues with similar structural motifs have been identified. Subsequent studies revealed that a series of aryl sulfonamides function as molecular glues, enhancing the interaction between the E3 ubiquitin ligase CUL4-DCAF15 (DDB1 CUL4 Associated Factor 15) and RBM39 (RNA binding motif protein 39), thereby promoting RBM39 degradation. Several aryl sulfonamides, including Indisulam, CQS, and E7820, have been evaluated in clinical trials as potential antitumor agents (Figure 6C) (Han et al., 2017; Uehara et al., 2017).
Molecular glue degraders have generated significant interest in the research community. To facilitate the discovery of molecular glue degraders targeting diverse proteins and binding modes, there is an urgent need for new screening methods and rational design strategies that transition from serendipitous discovery to systematic exploration. (1) Innovative Screening Technologies: Ebert et al. developed a method to mine correlations between the cytotoxicity of 4,518 clinical and preclinical small molecules and the expression levels of E3 ligase components across a wide range of human cancer cell lines. This led to the identification of CR8 as a molecular glue degrader that binds to CDK12–cyclin K, recruiting the DDB1–CUL4–RBX1 E3 ligase complex to ubiquitinate cyclin K (Figure 6D) (Słabicki et al., 2020; Kozicka et al., 2023). Similarly, through chemogenomic screening, Han et al. discovered HQ461, a novel molecular glue that binds the ATP pocket of CDK12 and promotes interaction with DDB1, triggering cyclin K degradation (Figure 6D) (Lv et al., 2020). (2) Optimization of Molecular Glue Design: Daniel et al. introduced a transposable chemical handle that can convert protein-targeting ligands into molecular degraders. Using the CDK4/6 inhibitor Ribociclib as a prototype, they identified a covalent handle capable of inducing CDK4 degradation. Further optimization led to the development of a “fumarate” handle, a universal modular chemical tool for generating molecular glue degraders (Figure 6D) (Toriki et al., 2023). Additionally, Rao et al. employed a dual-target, dual-mechanism design strategy to combine multiple mTOR inhibitors with E3 ligase ligands. After extensive screening, they discovered the bifunctional molecule YB-3-17, which selectively degrades GSPT1 while inhibiting mTOR (Figure 6D) (Liu Y. et al., 2024). These studies demonstrate the growing sophistication of molecular glue degrader design and expand their potential applications across various therapeutic targets.
3.2 Targeted protein degradation via lysosomeMore recently, new TPD strategies have been developed to hijack the lysosomal degradation pathway, the major degradation pathway independent of the proteasome. As a central “garbage disposal” mechanism within cells, lysosomes facilitate the breakdown of various biomacromolecules, including proteins, lipids, carbohydrates, and nucleic acids, through endocytic, phagocytic, and autophagic pathways (Saftig and Klumperman, 2009). The lysosomal degradation system comprises the endosome/lysosome and autophagy pathways, which operate both independently and synergistically to degrade intracellular and extracellular components. The endosome/lysosome pathway involves a series of membrane-bound compartments that mediate the processing of internalized material. Endocytosed substances are first incorporated into early endosomes, then transported through endosome carrier vesicles, late endosomes, and ultimately delivered to lysosomes for hydrolytic degradation (Ding et al., 2020). Autophagy, an evolutionarily conserved process, plays a critical role in maintaining cellular homeostasis by clearing unnecessary or damaged organelles and proteins via a lysosome-dependent mechanism. In this process, targeted organelles and proteins are encapsulated within double-membrane vesicles, known as autophagosomes (Zhao et al., 2022). These autophagosomes subsequently fuse with lysosomes, where their contents are broken down.
3.2.1 Autophagy-targeting chimeras (AUTACs)AUTAC is an innovative targeted protein degrader that harnesses the autophagy pathway to facilitate the selective degradation of intracellular proteins and organelle debris (Sakamoto et al., 2001). By inducing specific ubiquitin modifications, particularly K63-linked polyubiquitination, AUTAC molecules create a recognition signal that is effectively processed by the selective autophagy machinery. This K63 polyubiquitination is distinct from the more common K48-linked ubiquitination, which typically signals for proteasomal degradation (Figure 5C) (Takahashi et al., 2019). Structurally, AUTACs are heterobifunctional molecules comprising three key components: a degradation tag, which is often a guanine derivative responsible for initiating ubiquitination; a flexible linker that connects the degradation tag to the ligand; and a ligand warhead that selectively binds to the POI. Following binding, the targeted cellular components are sequestered into autophagosomes, which then fuse with lysosomes for degradation in the presence of lysosomal hydrolases. Despite the promise of AUTAC technology in drug discovery, significant gaps remain in understanding the detailed mechanisms underlying its efficacy, including the molecular interactions involved in the recognition and processing of ubiquitinated targets (Li et al., 2021). Currently, there are no clear examples of AUTACs in clinical trials, with research primarily focused on early preclinical assessments.
In 2019, Arimoto’s group introduced the concept of AUTAC, drawing inspiration from the antibacterial autophagy process. Since then, molecules of AUTACs 1–4 (Figure 7A) have been successfully employed to degrade various proteins, including methionine aminopeptidase 2 (MetAP2), FK506-binding protein (FKBP12), BET family proteins, translocator protein (TSPO), and others, alongside mitochondrial components, showcasing their remarkable degradation capabilities. Subsequent investigations have indicated that the degradation of mitochondria can trigger the elimination of additional pathogenic proteins (Takahashi et al., 2019). In 2023, the group conducted a SAR study, substituting L-Cysteine with pyrazole in the design of second-generation AUTACs. This modification resulted in a substantial enhancement of activity, with the tt44 (Figure 7A) of second-generation AUTACs achieving up to a 100-fold increase in potency compared to first-generation AUTACs, which were effective at concentrations of 10 μM (Takahashi et al., 2023). More recently, Schmitzer and colleagues developed an AUTAC-Biguanide compound (Figure 7A) by incorporating a biguanide functional group as a targeting moiety linked to the guanine scaffold. This strategic addition enables the selective targeting of the degradation process toward mitochondria. Notably, AUTAC-Biguanide exhibits superior antiproliferative properties compared to metformin and demonstrates enhanced selectivity for cancer cells (Vatté et al., 2024). This body of research underscores the potential of AUTAC molecules to effectively degrade cellular organelles, including damaged mitochondria, paving the way for novel therapeutic strategies.
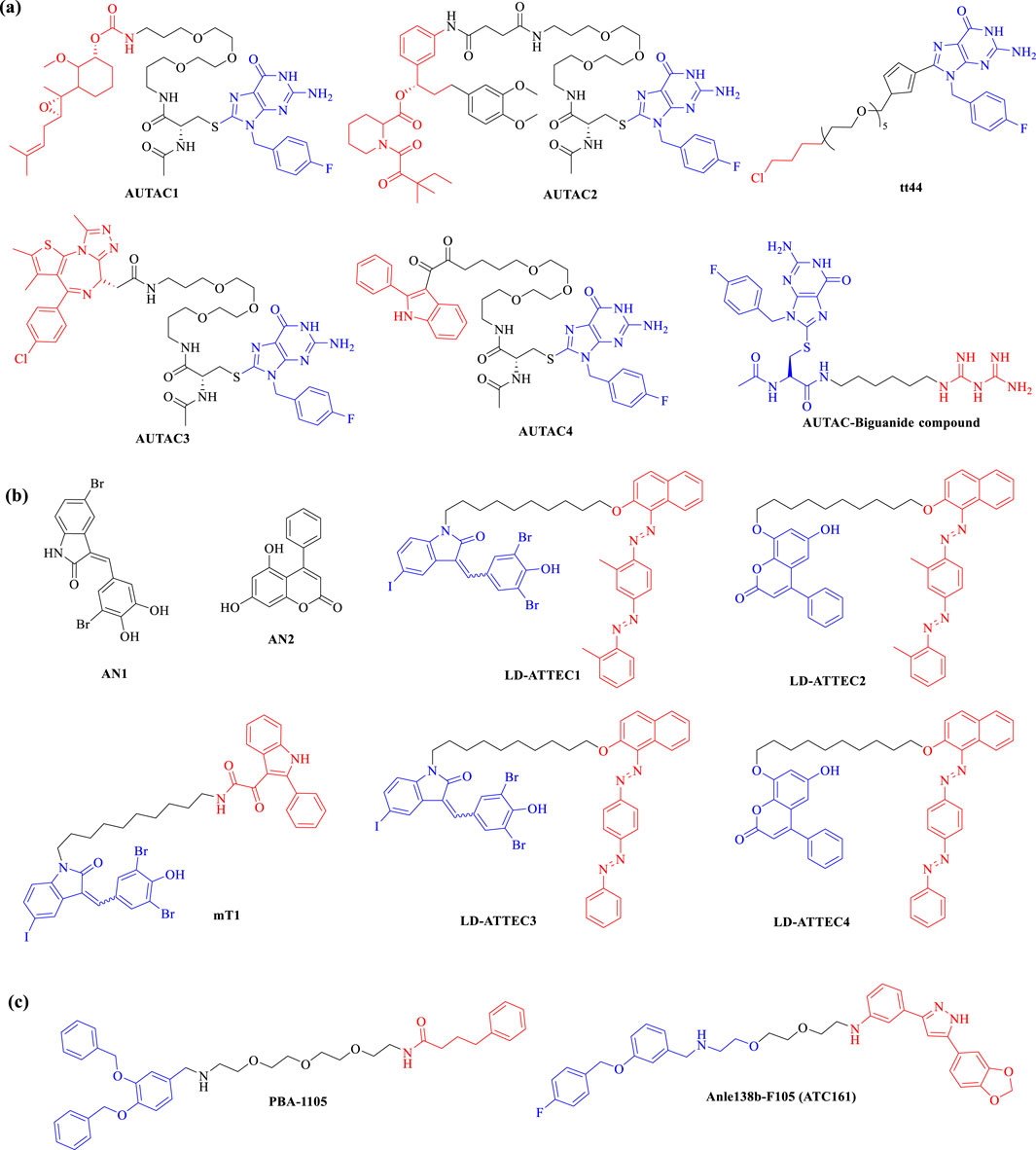
Figure 7. Chemical structures of lysosome-based degraders. Representational (A) AUTACs, (B) ATTECs and (C) AUTOTACs currently in preclinical trials. Degradation tag and proteins of interest (POI) warheads are respectively marked in blue and red.
3.2.2 Autophagosome-tethering compounds (ATTECs)ATTEC was first introduced in 2019 by Professor Boxun Lu of Fudan University (Li et al., 2019). Unlike PROTACs and AUTACs, which rely on ubiquitination for protein degradation, ATTEC molecules operate independently of this modification. Instead, ATTEC compounds function by tethering the POI to LC3, effectively directing disease-related targets to phagocytes for degradation (Figure 5D). This unique mechanism provides broad applicability across various disease contexts and target types (Zhang C. et al., 2024; Li et al., 2019). Distinct from PROTACs and AUTACs, which are characterized by two ligands connected via a linker, ATTEC molecules do not utilize a linker and can be likened to “molecular glue.” This innovative design allows for the precise and efficient recruitment of target proteins. In recent years, a series of novel ATTECs have been developed as heterobifunctional small molecules, enhancing their functionality. Given the diverse range of autophagic substrates, ATTEC technology holds significant promise for extending the applicability of protein degraders to non-proteinaceous biomolecules and organelles. This adaptability may pave the way for new therapeutic strategies targeting a wider array of biological processes, potentially transforming treatment approaches for various diseases.
In 2019, the Lu research group introduced ATTEC technology, developing ATTEC molecules capable of binding both the key autophagy protein LC3 and the mutant huntingtin protein (mHTT). This pioneering study demonstrated that molecules AN1 and AN2 (Figure 7B) effectively degrade mHTT in both cellular and in vivo animal models, successfully rescuing phenotypes associated with Huntington’s disease (Li et al., 2019). Building on this initial work, Lu and colleagues expanded the application of ATTEC technology by designing small molecules that target lipid droplets (LDs), which are implicated in various metabolic disorders such as obesity, type 2 diabetes, liver steatosis, and atherosclerosis. These novel ATTECs (LD-ATTECs C1-C4, shown in Figure 7B) bind to both LC3 and LDs, achieving near-complete clearance of LDs and rescuing LD-related phenotypes in cell cultures as well as in two independent mouse models (Fu et al., 2021). The proof-of-concept findings underscore the potential of ATTECs to facilitate the degradation of LDs. Most recently, Lu’s team has developed new bifunctional ATTEC (mT1, shown in Figure 7B) that simultaneously bind to the outer mitochondrial membrane protein TSPO and the autophagosome protein LC3. This innovative approach enhances the engulfment of damaged mitochondria by autophagosomes, promoting their subsequent autophagic degradation (Tan et al., 2023). This study not only confirms the ability of ATTECs to degrade organelles but also presents promising new strategies for intervening in mitochondria-related disorders. Conceptually, this approach could be extended to target a range of other proteins and non-protein entities, broadening the therapeutic potential of ATTEC technology.
3.2.3 AUTOphagy-TArgeting chimeras (AUTOTACs)In 2022, Kwon’s group introduced AUTOTACs, an innovative degrader technology that degrades UPS-resistant misfolded proteins and their oligomeric/aggregated species (Ji et al., 2022). AUTOTACs directly bind to the ZZ-type zinc finger (ZZ) domain of sequestosome 1 (SQSTM1)/p62, eliminating the need for polyubiquitination. This bifunctional molecule comprises two key components: a ligand that targets the POI and a ligand that directs the molecule to the autophagy pathway via the p62 protein (Figure 5E). By effectively linking these components through a flexible linker, AUTOTACs facilitate the simultaneous degradation of the target protein while enhancing autophagic flux. This dual functionality not only improves the efficiency of protein degradation but also promotes cellular health by ensuring the clearance of damaged or unnecessary proteins through autophagy, offering a promising strategy for therapeutic interventions in various diseases.
In 2022, the research team led by Yong Tae Kwon developed AUTOTAC compounds that features a module specifically designed to interact with the ZZ domain of p62, alongside a module targeting the POI. Both in vitro and in vivo studies demonstrated that these AUTOTACs (e.g., PBA-1105, Anle138b-F105, shown in Figure 7C) effectively targets the androgen receptor (AR) in prostate cancer cells and methionine aminopeptidase 2 (MetAP2) in glioblastoma cells, showcasing its powerful degradation capabilities (Ji et al., 2022). Building on this success, in 2023, Kwon’s group synthesized a series of AUTOTAC molecules that bind both p62/SQSTM1 and α-synuclein aggregates, which are implicated in the progression of Parkinson’s disease. One notable compound, ATC161 (Figure 7C), is an oral drug characterized by favorable pharmacokinetic and pharmacodynamic profiles. It selectively induces the degradation of α-synuclein aggregates in both in vitro and in vivo models (Lee et al., 2023). This research underscores the potential of the AUTOTAC platform for advancing drug discovery in the context of proteinopathies and related diseases.
3.3 Challenges of targeted protein degradation in drug discoveryTPD has emerged as a groundbreaking modality in drug discovery over the past 2 decades, with PROTACs and molecular glues representing the most advanced technologies in this field. In recent years, lysosome-based degrader technologies have begun to gain traction, significantly expanding the range of disease-related targets that can be addressed, thereby offering new strategies for clinical treatment. However, the development of lysosome-based TPD technologies remains in its infancy compared to the more established PROTAC and molecular glue approaches. While multiple PROTAC compounds have shown great promise in clinical trials, their development is not without challenges. First, synthesizing PROTACs requires not only the identification of a suitable ligand for the protein of interest but also converting that ligand into a functional PROTAC, which can be a time-intensive process demanding considerable expertise in synthetic and medicinal chemistry. Additionally, PROTACs often struggle with issues related to cell permeability and oral bioavailability, primarily due to their complex chemical structures. Furthermore, traditional methodologies do not provide a comprehensive understanding of the mechanisms of action of PROTACs, necessitating the implementation of complementary technologies for thorough evaluation. In contrast, molecular glues are generally smaller and more drug-like, which enhances their potential for clinical application. However, significant challenges remain, particularly during the later stages of optimization. While lysosome-based TPD technologies are still emerging, further research is essential to elucidate the molecular mechanisms and SAR of AUTACs, ATTECs, and AUTOTACs, and to establish their fundamental design principles. Despite these hurdles, TPD technologies hold immense potential to not only serve as powerful tools for biomedical research but also to enrich the medicinal chemist’s toolkit. With focused research and development efforts, these technologies could substantially broaden the spectrum of degradable targets, paving the way for exciting new avenues in therapeutic discovery.
4 DNA-encoded libraries (DELs) technologyDELs have emerged as a revolutionary tool in drug discovery, offering significant advantages in the identification of potential therapeutic candidates. Initially conceptualized by Brenner and Lerner in 1992, who proposed a theoretical framework for utilizing DNA to encode synthetic peptides (Brenner and Lerner, 1992), DEL technology saw practical application when Liu and Gartner employed DNA-templated synthesis to create small molecules with diverse chemical structures in 2001 (Gartner and Liu, 2001). DELs consist of combinatorial libraries of drug-like molecules, each uniquely barcoded with a DNA sequence that encodes the structure of the attached library member (Clark et al., 2009; Potowski et al., 2021). This innovative approach enables the preparation of libraries with unprecedented diversity and facilitates efficient screening for ligands that bind to specific protein targets. Traditional methods of synthesizing and screening individual molecules are often costly and complex, requiring substantial quantities of target protein, well-established bioassays, and intricate logistical arrangements. In contrast, DEL selections can be performed in just a few days, utilizing only tens of micrograms of protein and picomole amounts of DELs, along with next-generation DNA sequencing (NGS) technologies (Matsuo et al., 2023) (Figure 8). Compared to conventional compound libraries, DELs offer higher density, improved stability, and reduced costs, thereby opening up new avenues for the design and synthesis of complex molecules. This capability not only enhances the efficiency of the drug discovery process but also expands the range of chemical diversity that can be explored, ultimately contributing to the development of more effective therapeutics.
留言 (0)