
記住我







Cancer remains one of the leading causes of morbidity and mortality worldwide [1]. As a complex cascade, the progression of malignant tumors is shaped by a combination of intrinsic and extrinsic factors [2]. The carcinogenic roles of certain viruses, including human papilloma virus (HPV), hepatitis C virus (HCV), and Epstein–Barr virus (EBV), have been firmly established through both animal models and clinical trials. Accumulating evidence further highlights the potential involvement of bacteria and fungi in the process of malignant transformation [3, 4].
The human gastrointestinal tract harbors a vast community of over 100 trillion bacteria [5], which play a crucial role not only in normal development and physiological homeostasis [6, 7] but also in the initiation and progression of various cancers. Notably, Helicobacter pylori, the most well-known driver of gastric cancer [8], infects up to 43.1% of the global population [9]. Similarly, Escherichia coli, a pathogenic bacterium that establishes a symbiotic relationship with humans from infancy, has been shown to induce DNA damage in intestinal epithelial cells (IECs) [10]. Furthermore, in macrophage-deficient mouse models, Candida tropicalis has been observed to recruit myeloid-derived suppressor cells (MDSCs), thereby promoting the development of colorectal cancer (CRC) [11].
Gut dysbiosis, characterized by an imbalance between probiotic and pathogenic microorganisms, has been increasingly associated with the development of gut precancerous lesions, including irritable bowel syndrome (IBS) [12], inflammatory bowel disease (IBD) [13], colorectal polyps [14], and CRC [15]. Recent advances in the application of next-generation sequencing have significantly enhanced our understanding of the composition and functional dynamics of microbial community, shedding light on the intricate connections between the gut microbiome and the pathogenesis of extraintestinal cancers [16,17,18,19]. The gut microbiota profiles of cancer patients exhibit marked differences from those of healthy individuals. Moreover, accumulating evidence suggests that many microorganisms detected within tumor tissue may originate from the intestinal microbiota [20,21,22,23]. Despite these advances, the precise pathogenic mechanisms remain largely unknown, particularly regarding how the gut microbiota disrupts the intestinal barrier, disseminates through the circulatory system, colonizes distant organs, and ultimately exerts carcinogenic effects on parenchymal cells.
This review highlights recent advancements in understanding the close association between gut dysbiosis and tumorigenesis in extraintestinal organs, including the liver, breast, lung, and pancreas. A particular emphasis is placed on elucidating the molecular mechanisms by which the gut microbiota contributes to the pathological continuum from chronic inflammation and dysplasia to primary and metastatic tumors. Given the substantial evidence indicating that a significant proportion of intratumoral microbiota may represent translocated gut microbiota, we also explore their potential contributions to the establishment of the tumor microenvironment (TME). Additionally, we critically evaluate the advantages, limitations, and future directions of diagnostic technologies for detecting and characterizing extraintestinal tumors.
Relationship between the intestinal microbiota and extraintestinal cancersIntestinal barrier impairmentClinical evidence underscores that dysbiosis is a serious threat to the integrity of the gut barrier, a phenomenon temporally linked to the pathogenesis of various extraintestinal cancers [24]. The human body employs a sophisticated system to maintain gut homeostasis, which is essential for resisting the invasion of pathogenic microbiota and their detrimental metabolites during host-microbe interactions [25]. The gut barrier is a multifaceted structure comprising two primary components: (1) a physical barrier, which includes the mucus layer, IECs/intercellular junctions and endothelial cells of blood vessels; and (2) a functional barrier, primarily consisting of antibacterial proteins and intestinal alkaline phosphatase [25,26,27]. The physical barrier, also known as the mechanical barrier, plays a pivotal role in preventing the translocation of luminal microbiota and metabolites across the intestinal epithelium under homeostatic conditions [28, 29].
The outer mucus layer, predominantly formed by mucins, acts as the first line of defense, shielding epithelial cells from pathogens while fostering a symbiotic relationship with commensal microbiota [29]. Beneath this mucus layer lies a selectively permeable barrier consisting of a diverse array of epithelial cells, including enterocytes, enteroendocrine cells, Paneth cells, tuft cells, goblet cells, microfold cells, and intestinal stem cells [30, 31]. These epithelial cells are interconnected by specialized junctional structures such as tight junctions (TJs), adherens junctions, desmosomes, and gap junctions. TJs, located at the apical region of epithelial cells, constitute the majority of intercellular junctions and are predominantly composed of structural proteins like occludin, claudins, junctional adhesion molecules (JAMs), and tricellulin [32]. Additionally, functional proteins such as zonula occludens-1 (ZO-1), ZO-2, and ZO-3 regulate paracellular permeability, ensuring the selective passage of molecules and microorganisms to maintain intestinal barrier integrity [33,34,35].
The intestinal vasculature is lined by the gut vascular barrier (GVB), a specialized network of endothelial cells interconnected by TJs. This barrier is further supported by pericytes and enteric glial cells, ensuring the selective exchange of substances between the intestinal lumen and bloodstream [28, 36]. The integrity of GVB is maintained by specific proteins, including plasmalemma vesicle-associated protein-1 (PV-1), which plays a critical role in modulating barrier permeability and has been implicated in the progression of various diseases [37].
Bjarnason et al. first coined the term “leaky gut” to describe a pathological state of the intestine characterized by increased permeability, initially observed in individuals with excessive alcohol consumption [38]. This condition may enable the translocation of microbiota and/or harmful metabolites into the bloodstream, potentially triggering chronic immune responses and predisposing inflamed organs to carcinogenesis. Intestinal barrier damage can be categorized into endogenous and exogenous subtypes [39]. Lipopolysaccharide (LPS), also known as endotoxin, is primarily derived from the outer membrane of gram-negative bacteria in the gut and exhibits extensive biological activities. Even at low concentrations, LPS can elicit a potent inflammatory response by binding to Toll-like receptor 4 (TLR4) and its coreceptor myeloid differentiation protein 2 on immune cells. This interaction activates the transcription factor NF-κB, leading to the production of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and IL-1β [40, 41]. LPS disrupts intestinal barrier function by activating the TLR4/NF-κB signaling pathway, causing significant mislocalization of the ZO-1 protein [42, 43].
Furthermore, inflammatory cytokines like interferon-γ (IFN-γ) and TNF-α can exacerbate intestinal permeability through synergistic mechanisms, including the induction of IECs death and the internalization of transmembrane proteins [39]. IFN-γ also activates the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway, prolonging the NF-κB response, which reduces occludin expression and increase TJ permeability [44]. Meanwhile, TNF-α exerts its effects by activating myosin light chain kinase (MLCK) [45], resulting in cytoskeleton rearrangement, contraction of the perijunctional actomyosin ring, and ultrastructural alterations in TJs [46, 47].
Several external factors, including transient exposure to pathogenic microbes, alcohol consumption, drugs, and unhealthy dietary habits, can adversely affect intestinal permeability [48]. Pathogenic bacteria often utilize the type III secretion system (T3SS) to inject effector proteins into eukaryotic cells, disrupting the host cytoskeleton [49]. Salmonella, a common foodborne pathogen, is typically transmitted through contaminated food or water [50]. Mutations in the Salmonella SPI2 T3SS result in the formation of enlarged Salmonella-containing vacuoles within IECs. SPI2 T3SS effectors interact with the host’s microtubule network and intracellular transport systems, facilitating the apical-to-basolateral migration of these vacuoles. This process may contribute to increased intestinal permeability [51]. However, the precise mechanisms by which SPI2 T3SS effectors modulate the gut barrier, particularly their impact on epithelial TJ proteins, remain poorly understood and warrant further investigation.
Probiotic supplementation has demonstrated beneficial effects on intestinal barrier integrity across diverse age groups [52]. For example, feeding preterm infants formula supplemented with Bifidobacterium lactis (2 × 10⁷ CFU/g dry milk) for 30 days significantly decreases intestinal permeability [53]. Similarly, in middle-aged IBS patients (approximately 48 years old), the administration of Bifidobacterium longum BB536 and Lactobacillus rhamnosus HN001, combined with vitamin B6, has been shown to enhance gut microbiota diversity, decrease intestinal permeability, and alleviate clinical symptoms. Gut microbiota analysis revealed that the abundance of probiotic Lactobacillus/Bifidobacteria populations correlates positively with increased levels of short-chain fatty acids (SCFAs), both of which favor the maintenance of gut barrier integrity [54]. In elderly populations, a clinical trial demonstrated that supplementation with a probiotic mixture (2.0 × 1010 CFU of Lactobacillus paracasei HII01; 2.0 × 1010 CFU of Bifidobacterium breve; 1.0 × 1010 CFU of Bifidobacterium longum) promotes gut homeostasis [55]. These findings underscore the potential of probiotic-based interventions in repairing and preserving intestinal barrier function across different life stages.
Disruption of TJs is a key factor in alcohol-associated organ injuries [56]. Alcohol-induced dysbiosis activates monocytes and macrophages in the intestinal lamina propria to produce TNF-α, which binds to TNF-receptor I on intestinal epithelium and disrupts TJs through the activation of MLCK [57]. Antibiotic use, which affects individuals across all life stages, significantly disrupts the delicate balance of the gut microbiota and impairs host physiological functions [58, 59]. This disruption is akin to the “eradication” of microbial species, resulting in reduced microbial diversity and altered abundance within specific taxa, thereby indirectly compromising intestinal barrier function. Different antibiotics target specific microbial populations, impairing various components of the barrier. Antibiotic-induced disturbances reduce goblet cell function and mucus layer thickness in animal models. Alterations in pathogen-associated molecular pattern (PAMP) concentrations affect the secretory functions of IECs [60].
Recent studies have also explored the impacts of dietary fat on gut microbial composition, revealing that high-fat diets reduce the abundance of beneficial microbes that maintain intestinal barrier integrity while promoting the growth of potentially detrimental microbes [61, 62]. These alterations may negatively impact barrier function through metabolic pathways, such as the upregulation of lysophosphatidylcholine (LPC) and lysophosphatidic acid (LPA) [63]. In mice with intestinal epithelial cell-specific deficiency of fucosyltransferase 2 (Fut2), gut microbiota diversity is significantly reduced, contributing to increased LPC production. Both in vivo and in vitro experiments have demonstrated that LPC not only significantly reduces the number of mucus-secreting goblet cells but also inhibits the expression of TJ proteins such as ZO-1 and occludin [64].
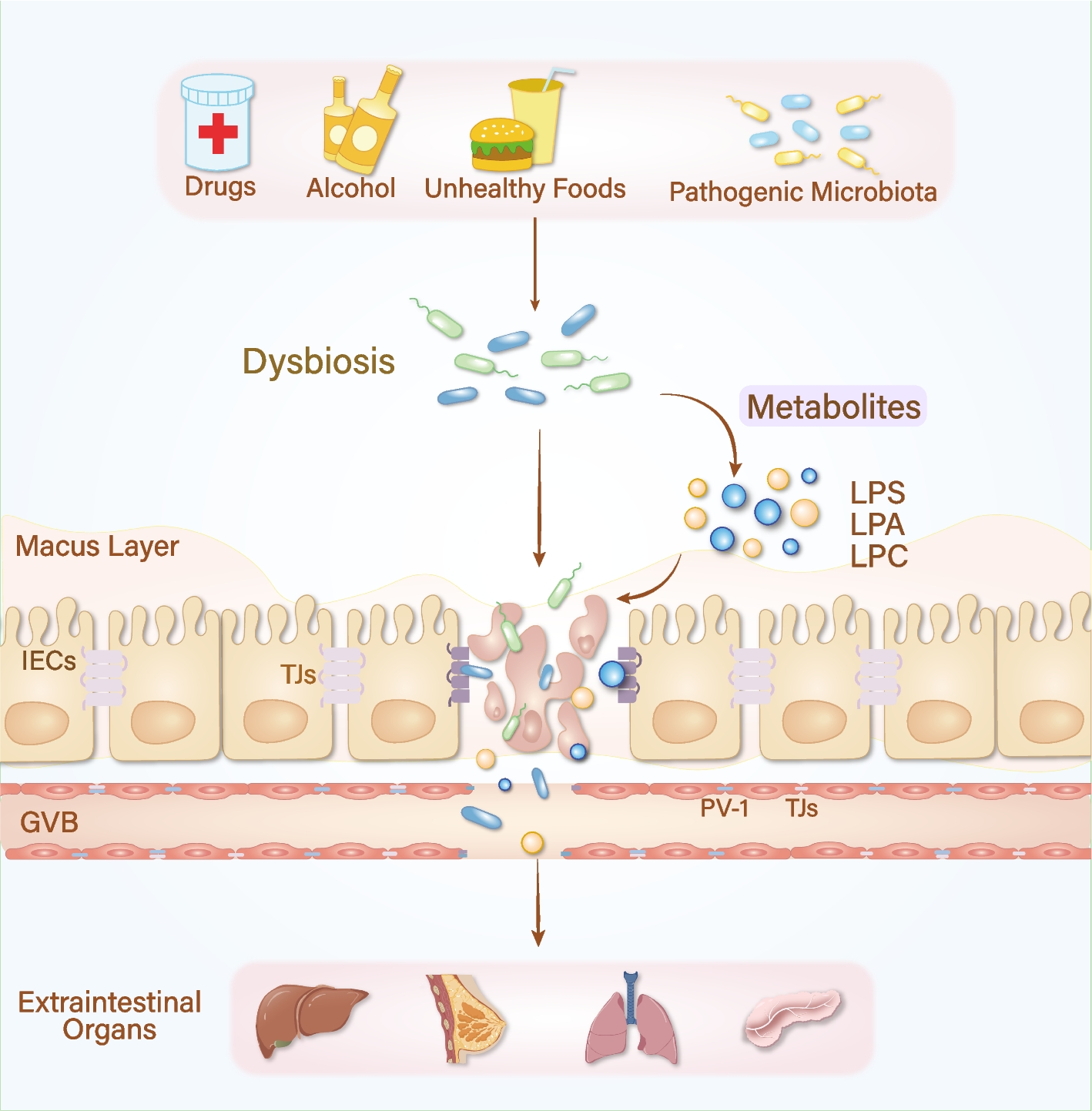
Given the profound influence of diet on the host microbiota, researchers have analyzed the diversity of food-associated microbiomes and integrated these findings with 19,833 human metagenomes [65]. It revealed significant overlap at the species and strain levels, highlighting the potential for therapeutic applications of food-derived microbiomes in clinical practice. Dysbiosis is influenced by a wide range of internal and external factors, resulting in diverse and complex mechanisms that regulate intestinal barrier permeability. This, in turn, promotes the spread of chronic inflammation, a well-established risk factor for tumorigenesis, making intestinal barrier dysfunction a potential predictor of cancer progression (Fig. 1).
Fig. 1
Life patterns, dysbiosis, and extraintestinal cancer. Exogenous detrimental factors, including pharmaceutical agents, alcohol consumption, high-fat dietary habits, and external pathogenic microbial exposure, contribute to intestinal barrier dysfunction and initiate a pathological continuum from chronic inflammation to metastatic cancer in distant organs such as the liver, breast, lung, and pancreas
Clinical data indicate that up to 80% of patients with cirrhosis eventually develop hepatocellular carcinoma (HCC). This oncogenic progression, also termed the “leaky gut” cascade, is often accompanied by gut dysbiosis and increased intestinal permeability [16]. The compromised intestinal barrier allows pathogens and their metabolites to translocate from the intestinal lumen into the portal system, where they land on the liver and initiate a pathological continuum from hepatitis and fibrosis to cirrhosis and, ultimately, HCC [66]. Disruption of GVB represents a critical rate-limiting step in this cascade. Spadoni et al. demonstrated that excessive colonization by intestinal bacterial pathogens, such as S. typhi, induces GVB damage, characterized by upregulated expression of PV-1, thereby promoting the systemic spread of bacteria and their toxic metabolites [67, 68]. In support of this hypothesis, Bertocchi et al. reported that E. coli (C17 strain) employs a virulence factor-dependent T3SS system to attack GVB in CRC patients, facilitating its entry into the bloodstream and subsequent extravasation into the liver. These translocated bacteria recruit immune cells to establish a premetastatic niche, fostering the development of metastases [69].
Metabolic-associated fatty liver disease (MAFLD) is a leading cause of HCC [70]. Increased intestinal barrier permeability, driven by intestinal inflammation, significantly exacerbates liver damage. Cheng et al. provided compelling evidence that mice fed a high-fat diet combined with dextran sulfate sodium (DSS) treatment exhibited more severe hepatic steatosis, inflammation, and fibrosis compared to controls fed a high-fat diet alone [71]. These findings underscore the role of DSS-induced colitis in aggravating hepatic degeneration. Further investigation revealed downregulated expression of epithelial ZO-1 and claudin-1 in DSS-treated mice, which facilitated bacterial invasion. Increased macromolecular permeability and elevated expression of endothelial PV-1 indicated GVB damage, promoting bacterial translocation and accelerating the progression of nonalcohol-associated steatohepatitis in the liver [71]. These results generally suggest that intestinal barrier injury acts as a critical mediator of liver damage and serves as an important risk factor for HCC development.
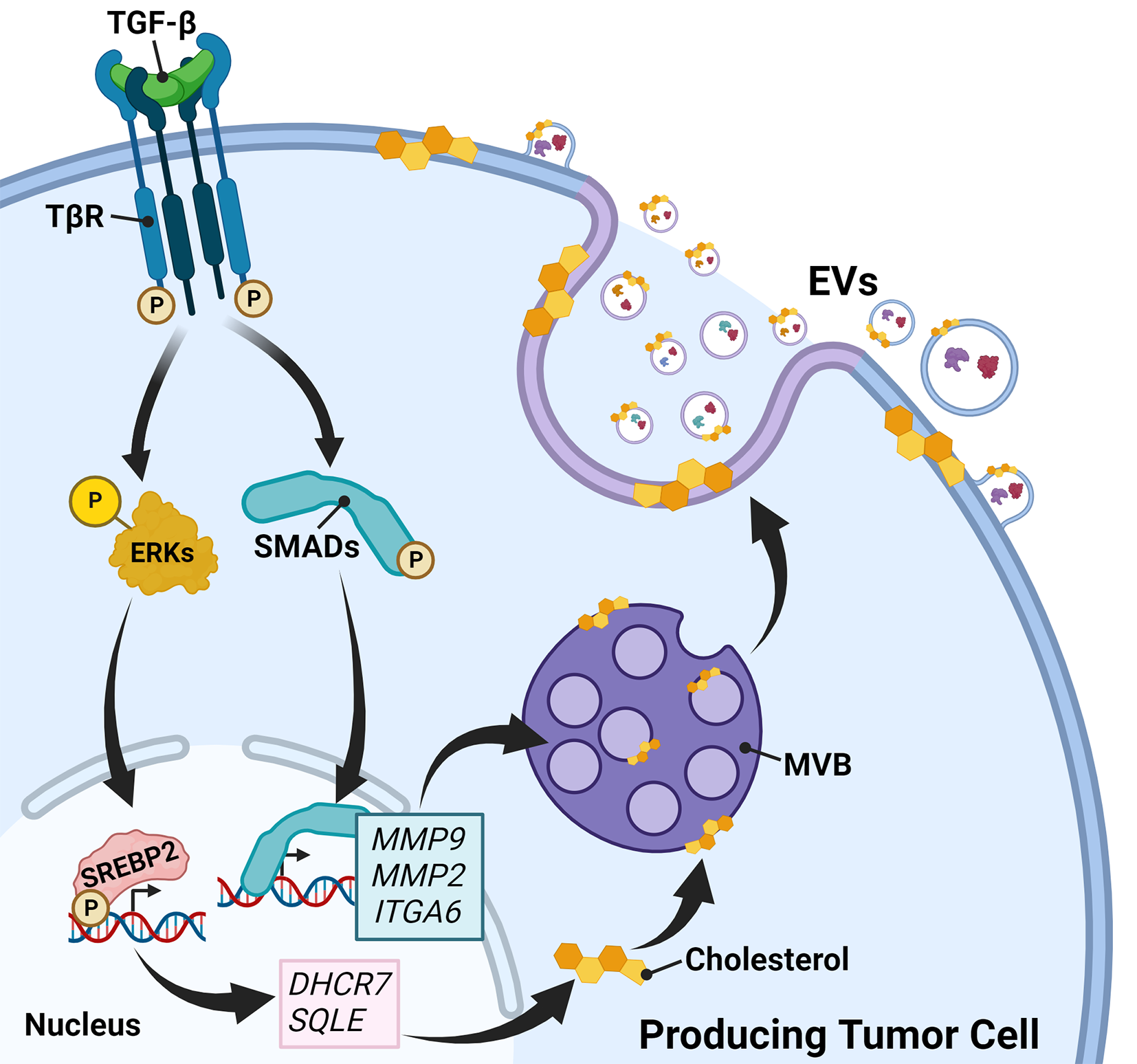
Intestinal barrier dysfunction also plays a significant role in the development of extraintestinal malignancies by modulating immune responses. When the intestinal barrier is compromised, gut-derived PAMPs and bacterial metabolites can enter the liver and subsequently activate TLRs, initiating protumorigenic immune responses [72]. Transplanting fecal microbiota from HCC patients into mice demonstrated that Klebsiella pneumoniae enhances the activity of macrophage-derived gelatinase in the colon, resulting in intestinal barrier dysfunction. This disruption facilitates the migration of K. pneumoniae to the liver, where its surface protein PBP1B binds to TLR4 on HCC cells. This interaction activates the TLR4 signaling pathway, ultimately driving the initiation and progression of HCC [73]. In chronically injured livers, TLR4 acts as a critical driver of HCC progression. Studies have shown that mice with genetically inactivated TLR4 exhibit a significantly lower incidence of HCC in the context of chronic liver injury [74]. TLR4 activation accelerates hepatic stellate cell (HSC) fibrosis via two distinct mechanisms. First, it enhances chemokine secretion (e.g., Ccl2 and Ccl5) by Kupffer cells, and second, it increases HSC sensitivity to transforming growth factor (TGF)-β, thereby accelerating early-stage HCC progression [75]. In advanced HCC, TLR4 expressed on hepatocytes provides survival signals to nontumorous liver cells, maintaining the viability of tumor precursor cells and facilitating further HCC development [74].
In addition, overexpression of TLR4 indirectly modulates the suppressive function of regulatory T cells (Tregs) and recruits Tregs through cytokine signaling [76], creating an immune exclusion barrier within the TME. This barrier prevents the infiltration of proinflammatory immune cells, thereby suppressing antitumor immune responses [76, 77]. Dysregulated activation of the TLR4/MyD88/NF-κB pathway has been identified as a significant risk factor for dysbiosis-associated extraintestinal tumorigenesis [78]. The leaky gut allows the accumulation of microbiota-derived LPS in the liver through the portal circulation. Upon binding to TLR4, LPS activates the MyD88/NF-κB signaling axis, leading to the upregulation of downstream proinflammatory cytokines such as IL-1β, IL-6, and TNF-α [79, 80]. Additionally, LPS directly enhances the migratory capacity of HCC cell lines by increasing the gene expression of IL-8 and TGF-β1 [81].
Dysbiosis also exerts detrimental effects on breast tissues, with accumulating evidence suggesting that infections induced by intestinal pathogenic microbiota can rapidly accelerate breast tumorigenesis [21], likely mediated by intestinal barrier disruption. The intestinal epithelial vitamin D receptor (VDR) is essential for maintaining gut and microbial homeostasis [82]. In conditionally deficient mice (VDRΔIEC), downregulation of ZO-1 indicates impaired TJ function and increased intestinal permeability, followed by significant alterations in gut bacterial abundance and metabolic activity [83]. Notably, compared with their WT littermates, VDRΔIEC mice develop more breast tumors and harbor gut-specific bacteria, including Streptococcus pyogenes, Streptococcus, Lactobacillus, Methylobacterium, and Atopobium, within their breast tissue. These findings suggest that intestinal bacteria infiltrate breast tissue through the compromised gut barrier, altering the local microenvironment and elevating cancer risk [83]. Treatment with the probiotic Lactobacillus plantarum improved breast VDR expression and restored colonic ZO-1 levels in VDRΔIEC mice, significantly reducing tumor incidence and size. This highlights the critical role of gut barrier dysfunction in breast cancer (BC) progression and underscores the potential of probiotics as a therapeutic strategy to mitigate cancer risk.
While the precise mechanisms by which gut microbiota translocate to the breast remain poorly understood, the immune system is likely a key mediator in this process. Pathogens may utilize macrophages as vectors to infiltrate deeper tissues, access blood vessels and lymph nodes, and subsequently disseminate to distant organs [84]. Under physiological conditions, the gut‒breast axis facilitates the transfer of maternal microbiota across the gut barrier to the breast, potentially via lymphatic drainage or through dendritic cells (DCs) and macrophages that transport microbial communities from mucosal tissues to lactating mammary glands [85, 86]. These observations suggest that disruptions in gut permeability and microbiota translocation can profoundly impact breast homeostasis. Understanding the mechanisms underlying microbiota dissemination is essential for developing strategies to protect breast health and prevent dysbiosis-related pathologies.
Active roles of the intestinal epitheliumCurrent research on dysbiosis has predominantly focused on the role of pathogenic bacteria in disrupting IECs and promoting malignant transformation. However, accumulating evidence indicates that IECs not only regulate the gut microbiota balance but also influence the homeostasis of distant organs. For instance, mutations in the Crumbs homolog 1 (Crb1) gene in mice lead to increased permeability in both the gut and retina [
留言 (0)