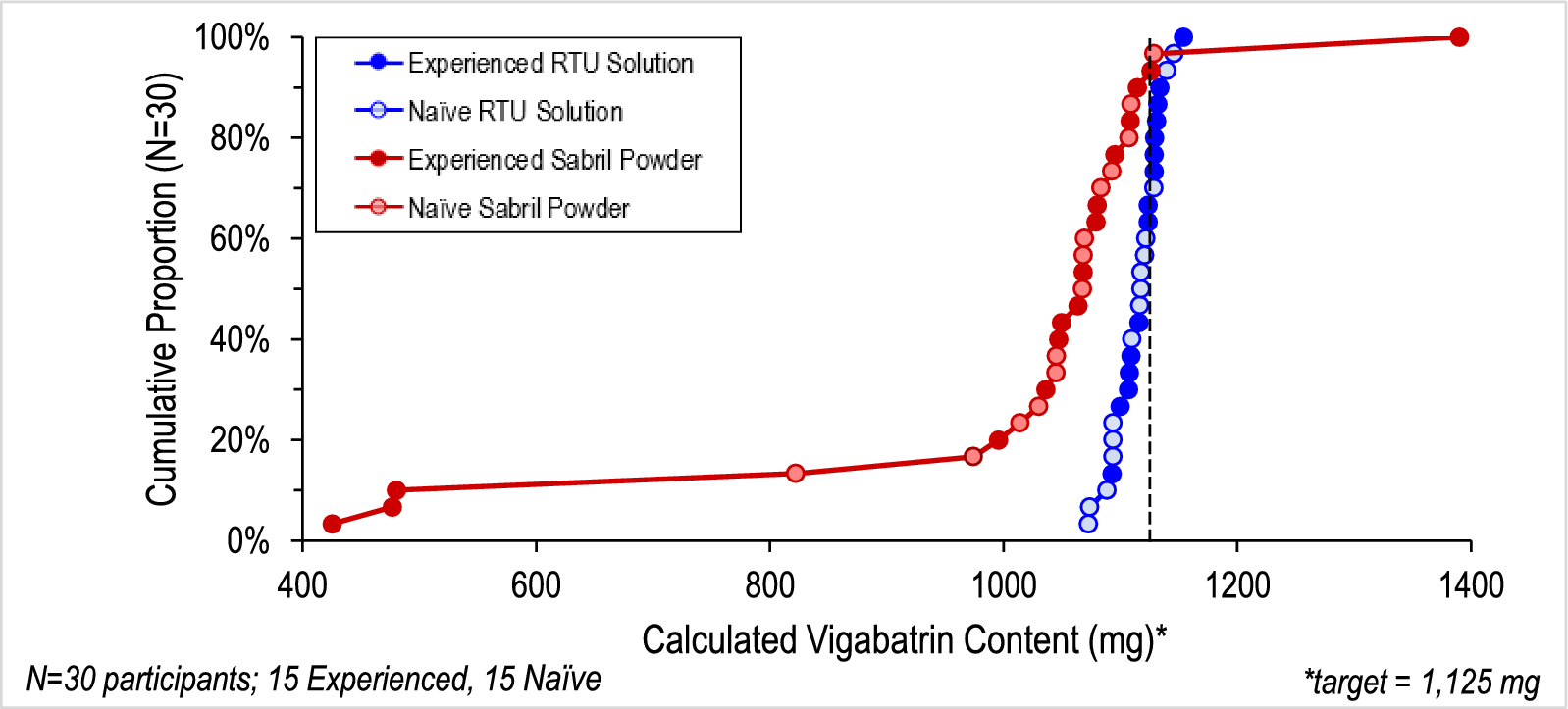
This study assessed the accuracy and variability of doses administered using a surrogate of VGB-RTU solution and vigabatrin powder. For the purposes of this study, the acceptable variability of target dosing was ± 10% of a target dose of 1125 mg (i.e., 1013–1238 mg). Additionally, the variability of dose delivered, along with variability and accuracy within subgroups (naïve and experienced users), was investigated.
A comparative, open label, crossover design was used during this study. Naïve participants (n = 15), representing caregivers of newly diagnosed infants, were either parents of young infants or adults of childbearing age who plan to have children who did not have prior experience using oral syringes (naïve) to provide medication. Per the protocol, experienced participants (n = 15) were caregivers who had administered vigabatrin in powder form to a child within the last 4 years; however, as a result of difficulties in enrolling caregivers of infants with IS, the study included two caregivers whose last experiences exceeded this window. The study was conducted at facilities in Boston, MA, and Chicago, IL. This nonclinical study was approved by a national Institutional Review Board, Castle IRB (Chesterfield, MO). The investigation was conducted in accordance with the 1964 Declaration of Helsinki and its later amendments, and was consistent with Good Clinical Practice and applicable regulatory requirements. Informed consent was obtained from participants prior to performing any study-related procedures.
The study excluded healthcare providers; those who had participated in market research studies within the past 6 months; had an occupation within market research, advertising, or a pharmaceutical/medical device company; or who were sharing a household with an individual working in those fields. Experienced participants averaged 40 years old (range 33–55 years old) while naïve participants averaged 28 years old (range 18–37 years old). Sixteen of 30 participants were male. The study was not powered sufficiently to detect differences between male and female caregivers.
The study was representative of a real-world situation that may be encountered by caregivers at home, and included a simulated kitchen environment equipped with all items required to successfully prepare the products per their labeling (i.e., measuring and preparation utensils, and liquids for reconstitution). A trained moderator, responsible for observing the participant’s interactions with each product and documenting study observations, was in the room with the participant. The participant could sit or stand (participant’s choice) at a counter or table to perform the simulated-use product preparation tasks.
VGB-RTU solution was provided in multidose 150-mL bottles with tamper-resistant foil seals and child-resistant caps. The VGB-RTU solution in this study, per FDA request, did not contain vigabatrin. This surrogate VGB-RTU solution, however, mirrored the physicochemical and viscosity characteristics of VIGAFYDE™. Since the concentration of vigabatrin in a ready-to-use solution is controlled by the approved manufacturing process and release testing rather than by the participant’s ability to prepare a correctly concentrated solution, the theoretical dose of vigabatrin is proportional to the volume of solution delivered. Per the proposed labeling, a syringe adapter and reusable 6-mL syringe with 0.25-mL graduations were supplied to each participant. Commercial VIGAFYDE™ solution is sweetened with a non-caloric sweetener (sucralose) and contains a mild mint flavor.
SABRIL® powder for oral solution was provided in packets containing 500 mg vigabatrin, each of which must be diluted by the caregiver with 10 mL of water (to produce a vigabatrin concentration of 50 mg/mL) prior to drawing up the prescribed dose using the provided 3-mL or 10-mL oral syringes. SABRIL® powder contains no sweetener nor taste masking agents.
VGB-RTU solution and vigabatrin powder were evaluated through simulated use scenarios (including analytical analysis). This crossover study required participants to prepare both products. If participants had previously used vigabatrin powder, this scenario was performed first. The order in which naïve participants performed each scenario was conducted in accordance with the randomization schedule. Participants were provided with the FDA-approved labeling for SABRIL® powder for oral solution, and labeling similar to that approved for VIGAFYDE™, and asked to provide a single dose of 1125 mg. No verbal instructions were provided, and the observers were not allowed to answer any questions. A total of 2 h was allocated for the participants to complete the tasks required to prepare both products. Participants delivered doses into a sample collection bottle to simulate the mouth of an infant. Actual infants were not included in this study. Each total delivered dose was weighed by a trained analyst with a calibrated Mettler Toledo XS6002S top loading analytical balance (precision ± 0.05 g; CV < 0.10%), and the weights were recorded to the hundredths place. The volume of the delivered dose was calculated on the basis of the density of the solution (VGB-RTU solution = 0.9976 g/mL, reconstituted vigabatrin powder = 1.000 g/mL).
Since the concentration of vigabatrin in VIGAFYDE™ solution is controlled by the manufacturer, the theoretical dose of vigabatrin administered (mg) using the VGB-RTU solution was calculated using the theoretical concentration of vigabatrin (100 mg/mL) multiplied by the volume of solution delivered. The dose of vigabatrin from solutions of vigabatrin powder in milligrams was calculated by multiplying the volume of solution delivered times the concentration of each prepared vigabatrin solution (in mg/mL) determined using a validated high-performance liquid chromatography (HPLC) assay. At least two control samples prepared by a trained analyst were interspersed in the randomization matrix each day, and were provided along with the study samples to the testing laboratory. The identities of the control samples were blinded to the laboratory personnel and were used to verify that the analyses performed by HPLC remained in a state of control throughout the study.
Study personnel collected sample weights, performed sample blinding, and shipped duplicate sample and control sample aliquots to the testing laboratory in real time after each day’s session in Boston. Samples from Chicago were shipped to the blinding facility for processing (i.e., weights, blinding, and aliquot preparation) at the end of each day prior to shipping the blinded sample aliquots to the testing laboratory.
Findings were analyzed by experience level, sample weights, sample volumes, vigabatrin doses in milligrams, and vigabatrin concentrations in milligrams per milliliter.
Reconstituted solutions of vigabatrin powder were diluted and tested using a HPLC assay based on the USP assay method described in the Vigabatrin for Oral Solution monograph after verification of system suitability, linearity, accuracy, precision, range, standard stability, and sample stability. Daily HPLC sequences met system suitability. Linearity was demonstrated to be acceptable with a correlation coefficient of 0.999995. Accuracy demonstrated a mean recovery (n = 9, 3 levels) of 100.0% and a percent relative standard deviation (RSD) of 0.3% (recovery 99.0–101.0%, ≤ 1.0% RSD). Precision was 0.1% RSD (≤ 1.0%). The method was shown to be accurate, linear, and precise, with a range of 1.0–3.0 mg/mL corresponding to 50–150% of the sample concentration after a 25-fold dilution. If samples fell outside of the validated range, dilutions were adjusted prior to reanalysis to ensure that all responses fell within the linear range. Sample stability was verified in sample bottles for 14 days and Eppendorf tubes for 4 days when stored at ambient conditions.
Data was evaluated for normal distribution. Consistently for all outcomes measures, VGB-RTU solution outcome measures were found to be normally distributed, while measures within the vigabatrin powder group did not satisfy normality evaluation via plots or Shapiro-Wilks testing. As such, a regression analysis using a generalized linear model with log transformation (log-link) of dose delivered, while accounting for repeated subject crossover design and controlling for subject experience in administering vigabatrin powder, was used to transform the data (SAS Proc Genmod). After transformation, log-transformed outcome measures satisfied normality assumptions. Transformed data were used to assess the variability of dose delivered and percentage dose delivered. Differences in proportions of dose deviation greater than 2% and 5% were reported as raw percentages with p values from two-sided Fisher’s exact tests. Applied statistical tests were two-sided, with statistical significance set at α ≤ 0.05. Because the data for vigabatrin powder was skewed and highly variable, log transformation was required. As a result, the data set was determined to be insufficiently powered to detect differences in accuracy or variability between experienced and naive users.



留言 (0)