
記住我
Rodents are reservoirs for over 60 zoonotic pathogens, contributing to the global emergence and re-emergence of infectious diseases (Woolhouse and Gowtage-Sequeria, 2005; Luis et al., 2013). Representing 43% of mammalian diversity, rodents are integral to the animal-human interface in Bushbuckridge-East, South Africa, where their prevalence in homes reaches 76% (Huchon et al., 2002; Berrian et al., 2016). Studies in this area have connected rodent-borne zoonoses with acute febrile illnesses in humans, finding 9.5% of such patients positive for Bartonella spp. and showing exposure to Coxiella burnetii and Leptospira spp. in others (Simpson et al., 2018). Given the association of certain Bartonella species with rodents, monitoring these hosts is vital, because their role in transmitting pathogens in Bushbuckridge-East remains unclear. Prior research in the area identified Anaplasma phagocytophilum in Mastomys natalensis and Rattus tanezumi captured from urban and periurban areas using 16S rRNA and gltA gene sequencing, highlighting the need for continued surveillance (Kolo et al., 2020).
Mastomys natalensis and R. tanezumi, closely associated with humans, may transmit infections (Skinner and Chimimba, 2005; Bonwitt et al., 2017). Next-generation sequencing (NGS) has unveiled diverse bacterial communities across hosts (Greay et al., 2018), yet rodent microbiomes remain underexplored (Cohen et al., 2015; Razzauti et al., 2015; Rynkiewicz et al., 2015; Ge et al., 2018). Research in China and France focused on bacteria detected from rodent spleens (Razzauti et al., 2015; Ge et al., 2018), while studies in Israel and the US examined the microbiome of rodent blood and their ectoparasites (Cohen et al., 2015; Rynkiewicz et al., 2015). These studies emphasized the importance of understanding host and vector bacterial communities as tools for managing vector-borne diseases (Cohen et al., 2015; Rynkiewicz et al., 2015). Despite rodents’ role as reservoirs for tick-borne pathogens (Telfer et al., 2007), South Africa’s wild murid rodents’ pathogenic diversity is poorly documented, often limited to single-genus studies (Hatyoka et al., 2019a, 2019; Kolo et al., 2020).
Mastomys natalensis and M. coucha, morphologically similar multimammate mice, are prevalent in southern Africa’s natural and human environments (Skinner and Chimimba, 2005), inhabiting grain stores and homes (De Graaff, 1981). Mastomys coucha is the primary host for flea-borne Yersinia pestis the cause of bubonic plague (Davis, 1964), while M. natalensis is associated with the Lassa fever virus in West Africa (Skinner and Chimimba, 2005).
The aim of this project was to elucidate the bacterial pathogens present in the blood of Mastomys species captured from different habitats at the animal-human interface in the Bushbuckridge-East community by sequencing of near full-length 16S rRNA gene amplicons.
2 Materials and methods2.1 Ethics approvalThe study received approval from the University of Pretoria’s Faculty of Veterinary Science animal ethics committee (V105-15). Research permissions for trapping and transporting rodents were granted by the South Africa Department of Agriculture, Land Reform and Rural Development (DALRRD) (12/11/1/1, 12/11/1/1/6), and the Mpumalanga Tourism and Parks Agency (MTPA, B1/290/2016), in accordance with the Animal Diseases Act (Section 20, 1984).
2.2 Study area and sample collectionThe research was conducted at the human-livestock-wildlife interface in Bushbuckridge bordering Manyeleti Game Reserve. A total of 282 rodents were captured across urban/periurban (Gottenburg and Hlalakahle), rangeland (Tlhavekisa), and protected areas (Manyeleti) from 2014-2015 (Supplementary Figure S1). Morphological identification (Stuart and Stuart, 2001) and sex was recorded. Rodents were humanely euthanized using isoflurane, and blood was collected via cardiac puncture in EDTA tubes and on FTA ™ cards at the Hans Hoheisen Wildlife Research Station. Blood samples were transferred to the University of Pretoria’s BSL3 lab where DNA was isolated using a QIAamp® DNA Mini Kit and stored at -20°C. The study utilized 24 samples from Mastomys spp., and one Steatomys sample. Seven samples were from Gottenburg, while the other sites had six samples each.
2.3 Molecular typing of Mastomys speciesMastomys rodents, a cryptic species complex, require molecular methods for accurate identification (Bastos et al., 2005). Two mitochondrial regions: the cytochrome b (cytb) gene and the barcoding region of cytochrome c oxidase subunit 1 (COI) were used for identification. Amplification and sequencing were performed at the University of Pretoria and Genelethu labs (Johannesburg), with COI sequences deposited and accessible through the portal of the Barcode of Life database (BOLD) https://portal.boldsystems.org/ with record set code (ZTBP) and in Genbank with accession numbers (PQ814604-PQ814628). Cytb sequences were deposited in Genbank with accession numbers (PP790712-PP790735).
2.4 Collection of ectoparasitesRegulations of the South African Department of Agriculture, Land Reform and Rural Development, (DALRRD) required that whole body rodent carcasses must first be frozen at -80°C and stored at Hans Hoheisen Research Centre, Mpumalanga South Africa before transport to a BSL3 provincial laboratory at Stellenbosch, South Africa. Ectoparasites, including ticks, fleas, lice, and mites, were removed in the BSL3 laboratory and preserved in absolute ethanol for morphological identification, using established taxonomic information (Till, 1963; Tipton et al., 1966; Ledger, 1980; Segerman, 1995; Walker, 2000; Horak et al., 2018). All ticks and lice were identified. A subsample of mites (trombiculid and mesostigmatan) and only male fleas were identified.
2.5 PCR amplification and sequencingThe 16S rRNA gene (V1-V8 variable regions) was amplified from 25 rodent samples with primers 27F and 1435R (Supplementary Table S1), as described (Gall et al., 2016). PCR was conducted using barcoded primers (Supplementary Table S2), Phusion Flash® High Fidelity PCR Master Mix, and 100 ng of DNA, with three technical replicates (Gall et al., 2016). Anaplasma centrale DNA and PCR grade water were used as PCR controls. Cycling parameters included 98°C for 30s, 35 cycles at 98°C for 10s, 60°C for 30s, 72°C for 30s, and a final extension at 72°C for 10 min. PCR products, visualized on a 1.5% agarose gel were purified with the QIAquickPCR® purification kit, and sent to Washington State University for Circular Consensus Sequencing on the PacBio platform.
2.6 16S rRNA sequence analysisSequence data was processed using PacBio software, adhering to predefined size and precision parameters. Genus-level classification of reads utilized the RDP 16S classifier (Cole et al., 2009) and NCBI BLASTn analysis for sequence identification. BLASTn results were filtered using Microsoft Excel, using a 1275 bp length and 98% identity threshold (Gall et al., 2016). Sequences below this threshold were classified at the genus level, while higher matches were identified to the species level (Jones et al., 2010; Bonnet et al., 2014; Budachetri et al., 2014). Operational taxonomic units (OTUs) representing less than 1% of total sequences were categorized as ‘rare’ (Gall et al., 2016). Raw microbiome sequence data were deposited in the NCBI sequence read archive (SRA) with accession numbers SRX5967121-SRX5967145 (Supplementary Table S3, Supplementary Material). For Bartonella, 16S consensus sequences were extracted using CLC genomics workbench 9.5.1 (Qiagen) and aligned with GenBank entries. The Jmodel test 1.3 predicted GTR + I + G as the optimal model (Darriba et al., 2012) selected under the Bayesian information criterion (BIC), and phylogenetic analysis was conducted using the maximum likelihood method in MEGA 11 (Tamura et al., 2021).
Rodent blood microbial compositions were analyzed with the community ecology package vegan 2.5-2 (Oksanen et al., 2016) in R studio (R Core Team, 2013). Alpha diversity was assessed through rarefaction curves, determining mean bacterial species diversity across habitats. Principal component analysis (PCA) quantifying bacterial population similarities in rodent blood, was done using FactoMineR (Lê et al., 2008). Similar blood bacterial profiles resulted in clustering, while dispersion showed variability. The variables proximity on the PCA plane suggested positive correlations; while opposite positioned variables indicated negative correlations. Correlation coefficient (r) between variables and the dimensions (Dim) were considered significant with p-values <0.05. Nonmetric multidimensional scaling (NMDS) ordination compared bacterial population differences, using distance analysis in Phyloseq. A Phyloseq-generated heatmap visualized rodent blood OTU diversity and abundance. Permutational ANOVA (PerMANOVA), using vegan’s adonis function and Bray-Curtis index with 1000 permutations (Oksanen et al., 2016) tested habitat-based bacterial composition differences, with significance at pseudo F-associated p-values ≤0.05.
2.7 Bartonella gltA gene characterizationCharacterization of Bartonella spp. in rodents was done using primers Bart-EF and Bart-ER targeting the citrate synthase (gltA) gene yielding approximately 500 bp fragments (Bastos, 2007). The PCR products were sequenced, and sequences submitted to Genbank (accession numbers: PP831190-PP831198). Sequence alignment was performed with closely related sequences, identified through nucleotide BLAST search. Phylogenetic analysis utilized the Maximum Likelihood method, applying the best-fit evolutionary model in MEGA 11 (Tamura et al., 2021).
3 Results3.1 Molecular typing of Mastomys hostsAnalysis of the COI gene classified 10 out of 25 Mastomys specimens as M. coucha, one as Steatomys sp., and the remainder as M. natalensis. This was corroborated by cytb gene sequencing (Supplementary Table S2). Habitat distribution patterns indicated a prevalence of M. natalensis in urban and peri-urban settings, whereas M. coucha was more frequently encountered in communal rangelands and protected areas. Closer examination of the COI gene sequences from M. natalensis revealed minor variations, with individual specimens R31, R84, and R74 exhibiting single nucleotide differences at specific loci within a 650-nucleotide sequence. Greater genetic diversity was observed in M. coucha, with a 658-nucleotide sequence analysis showing single nucleotide variations among specimens R2, R12, R159, R177, and R179. Notably, R2 and R12 shared identical sequences, as did R159 and R177. Additionally, unique nucleotide substitutions were identified in specimens R5, R6, R11, R20, and R61, each at distinct positions.
3.2 EctoparasitesA total of 439 ectoparasites comprising 10 ticks (4 Haemaphysalis leachi/elliptica group, 4 Rhipicephalus simus/follis group and 2 Rhipicephalus microplus), 337 mites of which 5 were trombiculid mites, 61 fleas (mainly Xenopsylla brasilliensis at >30% prevalence) and 31 lice (21 Polyplax biseriata and 10 Holopleura intermedia) were collected from the rodents (Table 1).
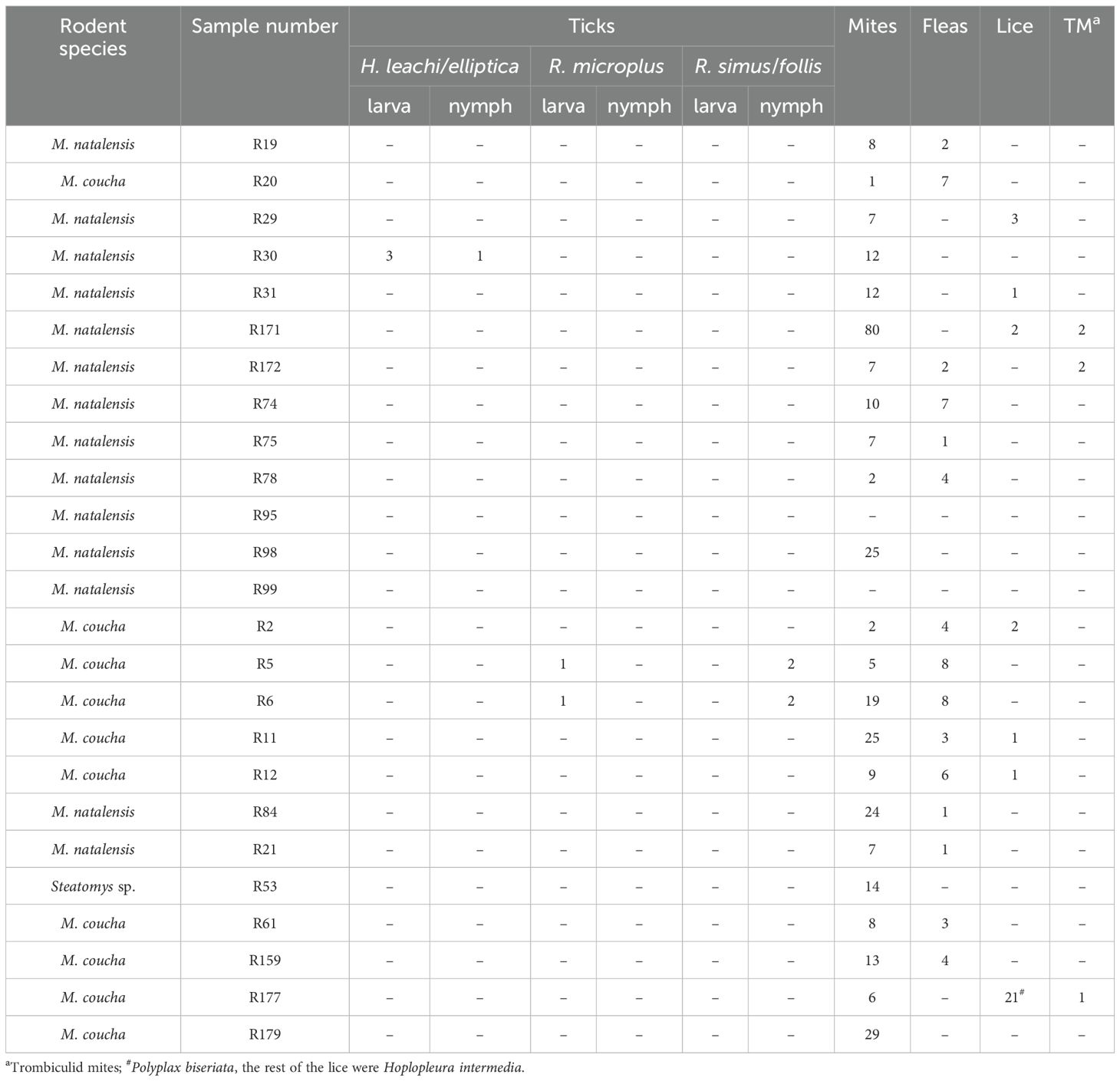
Table 1. Ectoparasites recovered from Mastomys spp. and Steatomys.
3.3 Barcoded 16S rRNA gene sequencing and statistical analysisSequencing of the barcoded 16S amplicons generated 65,059 bacterial sequences, with a mean of 2,602 reads per sample, confirming the capture of all OTUs to satisfy a rarefaction curve (Supplementary Figure S2). The species diversity, represented by a plateau in the rarefaction curve, indicated a well-sampled bacterial community. Analysis identified 17 OTUs across ten species and six genera, excluding rare and unclassified OTUs. Approximately 90.0% of the total reads (58,543 out of 65,059) were assigned to valid taxa. Bartonella grahamii was the most prevalent, forming 29.0% of the bacterial sequences, followed by Bartonella mastomydis at 23.0%. Bartonella spp. that fell below the 98% cut-off point made up 12.0% of the sequences. Other bacteria included Pseudomonas spp. (17.9%), Ochrobactrum spp. (7.2%), and Anaplasma spp. (0.5%), B. henselae (0.1%), Ehrlichia sp. (0.03%), and Coxiella burnetii (0.02%). Anaplasma spp. comprised 10 sequences of A. centrale, five sequences of A. phagocytophilum and two sequences of A. marginale. Rare and unclassified OTUs constituted 4.7% and 5.3% of the sequences. Predominant bacterial taxa are illustrated in Figure 1A.
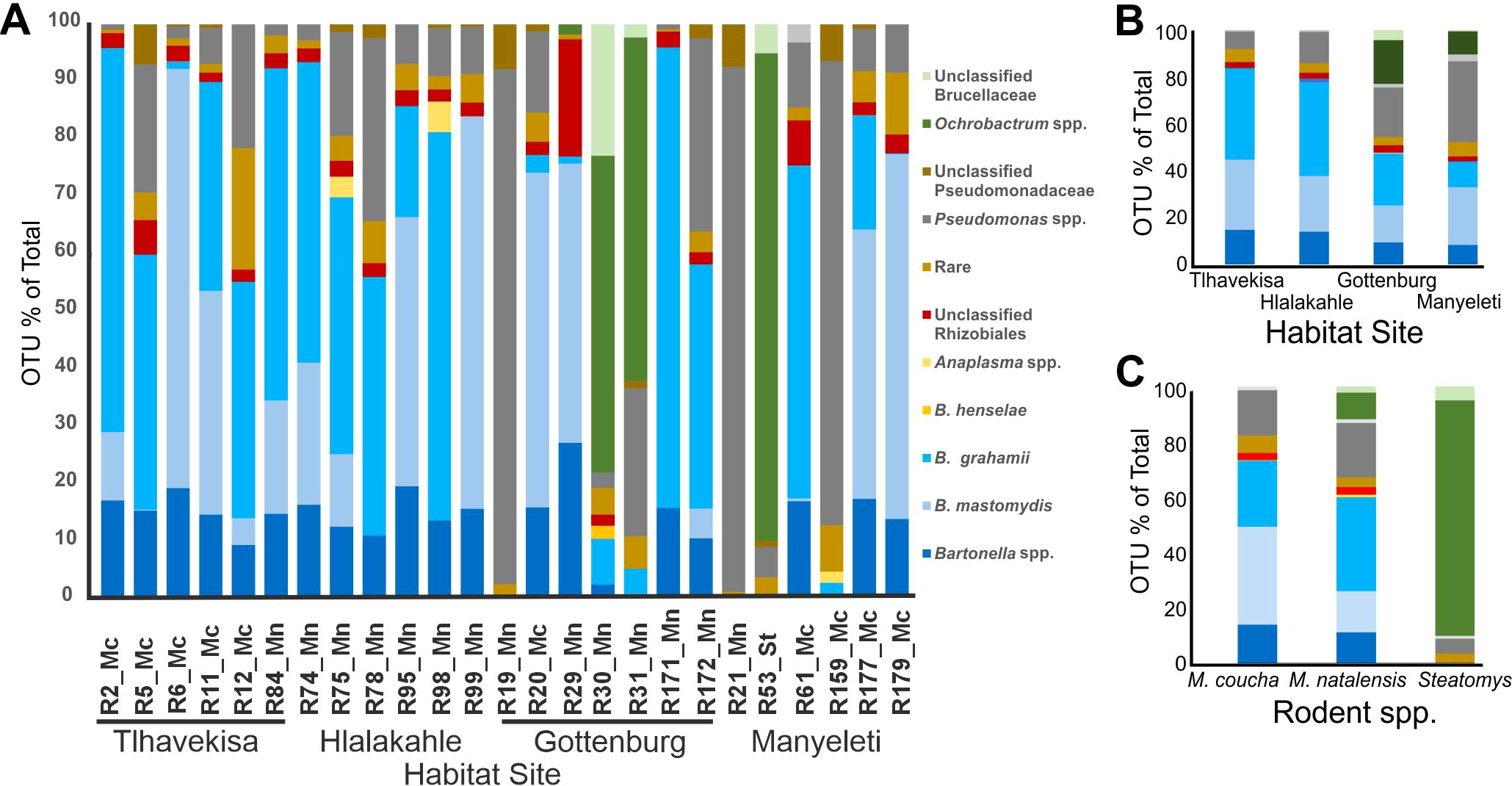
Figure 1. Relative abundance of major taxa of bacteria. (A) In individual rodents, six to seven rodents from each habitat area were sampled. Rodent species are linked to sample numbers (Mn abbreviation for M. natalensis, Mc for M. coucha and St for Steatomys). The village names are indicated under the rodent sample numbers. (B) In the blood of Mastomys spp. from different habitat areas: Hlalakahle and Gottenburg (urban/peri-urban area), Tlhavekisa (communal rangeland), and Manyeleti (protected wild reserve) and (C) In the blood of Mastomys coucha, M. natalensis and Steatomys sp.
3.3.1 OTUs by habitat areaA heat map of OTU diversity in rodent blood, grouped by capture location (Supplementary Figure S3) revealed high Bartonella spp. prevalence (~83%) in rodents from Hlalakahle (peri-urban) and Tlhavekisa (communal rangeland), contrasting with lower occurrences (~45%) in Gottenburg (peri-urban) and Manyeleti (wildlife reserve). Ochrobactrum spp. was detected in Gottenburg and Manyeleti specimens. Ehrlichia sp. was detected in R75 (Hlalakahle) and R172 (Gottenberg), while Coxiella sp. was exclusive to rodent R12 (Tlhavekisa). Anaplasma phagocytophilum was identified in Rodent R98 (Hlalakahle) while A. centrale and A. marginale were identified in R20 (Gottenburg). Manyeleti rodents exhibited the highest Pseudomonas spp. infection rates (~34%), influenced by significant burdens in rodents R21 and R159. The microbial profile of rodent blood, based on rodent capture sites, is depicted in Figure 1B.
3.3.2 OTUs by rodent speciesBased on rodent species, B. grahamii accounted for 23.9% and 33.9% of sequences in M. coucha and M. natalensis. Bartonella mastomydis was more prevalent in M. coucha at 35.4%, compared to 14.9% in M. natalensis. Unspeciated Bartonella were comparably lower, constituting 13.9% in M. coucha and 11.1% in M. natalensis. Notably, B. henselae was exclusively found in M. natalensis, albeit a minimal 0.2%. Pseudomonas spp. showed a higher presence in M. natalensis (19.7%) than in M. coucha (16.4%), and Steatomys (5.3%). Conversely, Ochrobactrum spp. dominated in Steatomys with 85.1% and was less common in M. natalensis at 9.6%. The microbial composition in the blood of M. coucha, M. natalensis, and Steatomys sp. is shown in Figure 1C.
3.3.3 Relative abundance of taxaMost Bartonella sequences had 99% identity to B. grahamii strain as4aup (CP001562), while a subset matched B. mastomydis (AY993936). Ehrlichia sequences in rodents R75 and R172 showed 98% identity to strains EH727 (AY309970) and Ehf669 (AY309969), as well as Ehrlichia sp. Tibet (AF414399) and E. chaffeensis (NR_074500). Anaplasma sequences in rodent R98 were 99% identical to several A. phagocytophilum strains, including the Dog2 strain (CP006618) and human strain HZ (CP000235). Ochrobactrum spp. detected in rodents from Gottenburg and Manyeleti, showed 99% identity to O. intermedium (JN613288 & KT696500), and O. pseuintermedium (DQ365922). Pseudomonas spp., with 98% identity to P. extremaustralis (NR114911), Pseudomonas sp. BFXJ-8 (EU013945) and P. fluorescens (CP015638), were found in all rodents except R29. Lastly, rodent R12 had sequences with 99% identity to C. burnetii strains Schperling (CP014563) and Namibia (CP007555).
3.3.4 PCA and NMDS analysesPCA showed 30.60% of the variation in the rodent bacterial communities clustered with Dim1, while 25.16% clustered with Dim2. Rodent R30 from Gottenburg was distinct, while R19 (Gottenburg), R21 and R159 (Manyeleti) showed similarities, clustering at the mid to bottom right. Other rodents, R6, R29, R31, and R53, varied, while the rest formed a main cluster (Figure 2A). The variables factor map (Figure 2B) showed that Pseudomonas spp. (R= 0.7), unclassified Pseudomonadaceae (R= 0.6), Ochrobactrum spp. (R=0.6), unclassified Brucellaceae (R=0.5), and B. henselae (R= 0.4) were positively correlated with Dim 1. Conversely, Bartonella spp. (R= -0.9), B. mastomydis (R= -0.6), and unclassified Rhizobiales (R=0.6) correlated negatively with Dim 1 and positively with Dim 2. R30 and R31 (Gottenburg), and R53 (Manyeleti) were associated with variables positively affecting Dim 1, while the main cluster was linked to variables influencing Dim 2. Finally, R19 (Gottenburg), R21 and R159 (Manyeleti) correlated with Pseudomonas spp. and unclassified Pseudomonadaceae.
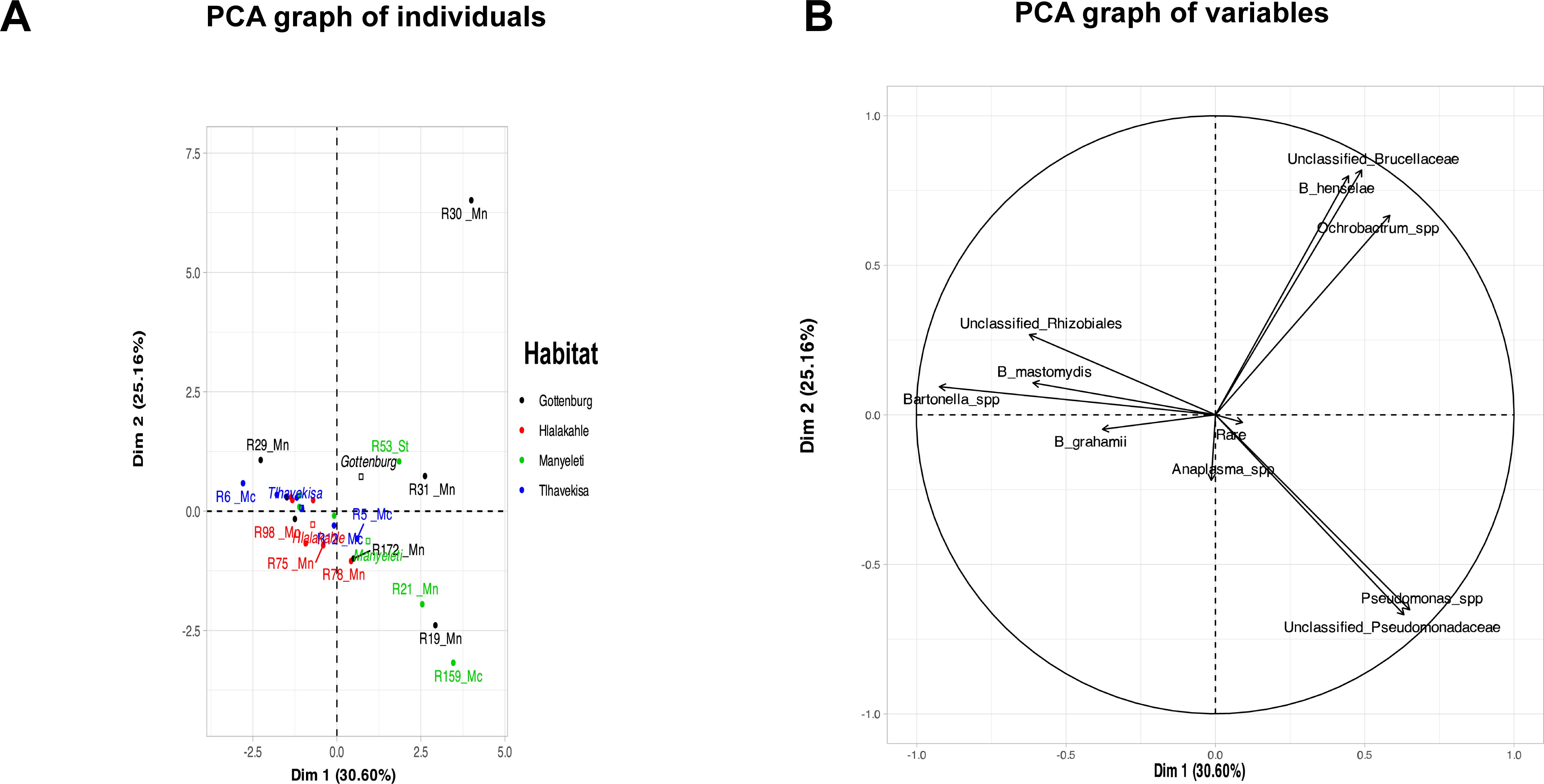
Figure 2. PCA plot analyzing the blood microbiome of rodents with the first two principal components explaining 30.60% and 25.16% of the variance, respectively. (A) Graph of individual rodents. Black dots represent samples from Gottenburg, red samples are from Hlalakahle, green samples are from Manyeleti and blue samples from Tlhavekisa. Clustering suggests similarities in bacterial profiles and dispersion indicates variability. Notably, R30 from Gottenburg is an outlier. Rodent species are linked to sample numbers (Mn abbreviation for M. natalensis, Mc for M. coucha and St for Steatomys). The position of habitat names reflects their significant contribution to the plot’s dimensions. (B) Graph of variables shows bacterial populations detected from rodent blood in the Bushbuckridge-East community.
The NMDS plot displayed variations in bacterial diversity of rodent blood across habitats and among M. coucha, M. natalensis, and Steatomys species (Supplementary Figure S3). Despite this, PerMANOVA revealed no significant differences in bacterial populations between rodents from Tlhavekisa (rangelands) and Hlalakahle (urban/per-urban area) (P = 0.1). The study’s small sample size limited our ability to draw statistically significant conclusions regarding the diversity of the bacterial blood microbiome of the rodents across the habitats.
3.4 Phylogenetic analysis of Bartonella spp.3.4.1 16S rRNA gene phylogenyFive rodents (R6, R11, R30, R75 and R179) from the different habitats had one or more Bartonella spp. co-infections. These sequences (8) were used for phylogenetic analysis of the 16S rRNA gene using the Maximum likelihood method (Figure 3A).
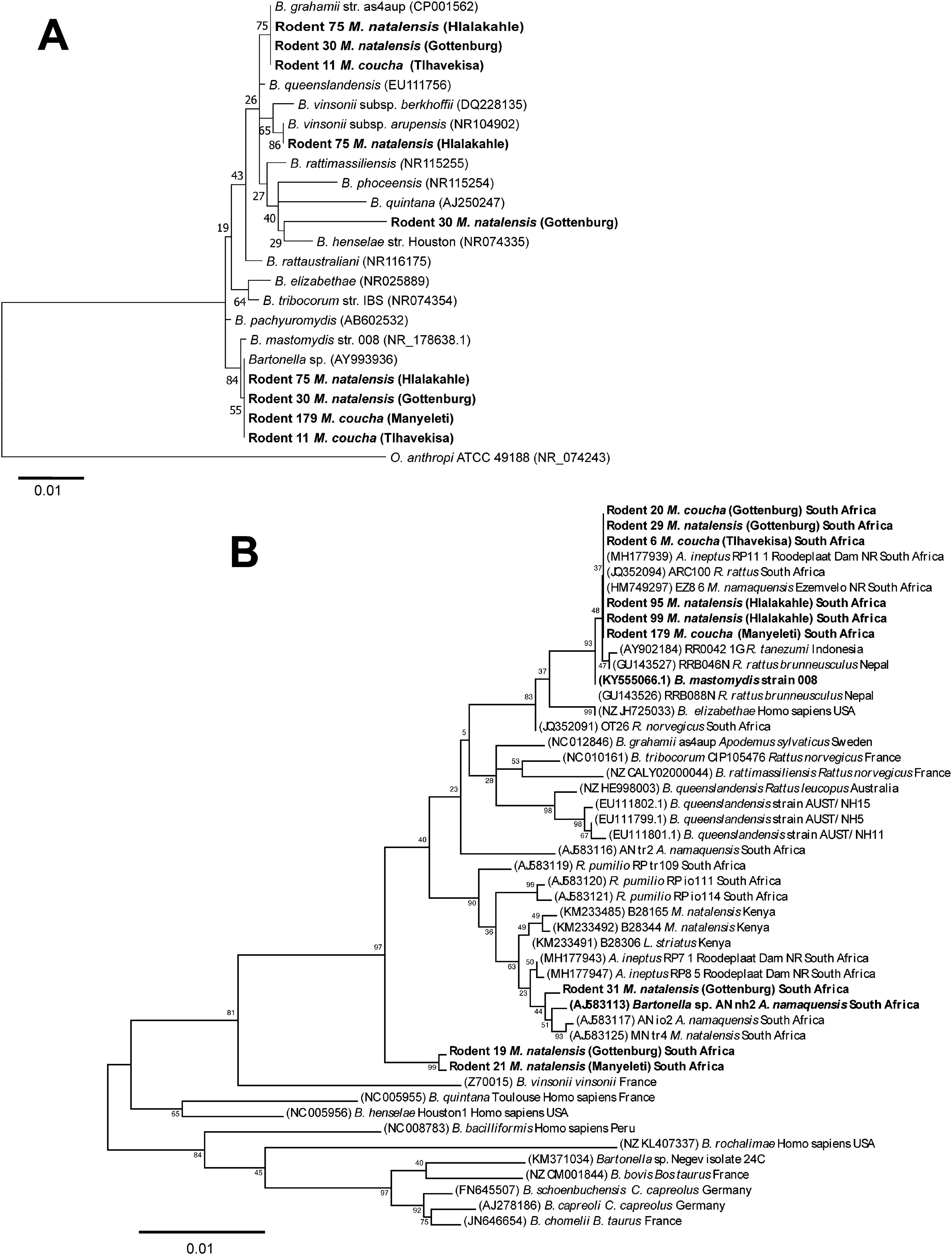
Figure 3. Maximum likelihood trees. (A) 16S rRNA gene tree inferred using the General Time Reversible (GTR) model of sequence evolution with G + I [G (0.1000), I (46.51% sites)]. Relationships between previously described Bartonella spp. and the Bartonella strains identified in the blood of five rodents from different habitat areas in the Bushbuckridge-East community (indicated in bold) are shown. Three rodents (11, 30 and 75) were co-infected with more than one Bartonella strains. Genbank accession numbers are given in parenthesis. The analysis involved 24 nucleotide sequences. There were 1309 positions in the final dataset. (B) gltA gene tree inferred using the Tamura 3-parameter + G (0.25) model of sequence evolution. Bartonella spp. identified in the blood of rodents from different habitat areas in the Bushbuckridge-East community and reference Bartonella sequences are indicated in bold, with rodents 6 and 179, common to both the 16S rRNA and gltA gene phylogenies. Genbank accession numbers are given in parenthesis. The analysis involved 47 nucleotide sequences. Bootstrap values from 1000 replications are indicated next to the relevant node. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site.
3.4.2 GltA gene phylogenySequence analysis of gltA gene sequences from R5, R11, R12, and R84 captured from Tlhavekisa, identified multiple Bartonella co-infections complicating species delineation. However, nine rodents yielded clean/unambiguous nucleotide sequences. Notably, six samples (R6, R20, R29, R95, R99, R179) from the various habitats were infected with B. mastomydis (Figure 3B). Potentially novel Bartonella were identified in M. natalensis: one in R19 and R21 (from Gottenburg and Manyeleti) which formed a distinct clade separate from known Bartonella spp., and the second in R31 (Gottenburg), closely related to Bartonella sp. AN-nh2 previously detected in Micaelamys namequensis (Pretorius et al., 2004). Sequences from R6 and R179 contributed to both 16S rRNA and gltA gene phylogenies.
4 DiscussionThis study identified a variety of bacterial species in the blood of Mastomys and Steatomys spp., predominantly Bartonella spp., at 64% of sequences. All rodents were infected, aligning with prior research that showed Bartonella’s prevalence in rodent blood and fleas in Israel and the US (Cohen et al., 2015; Rynkiewicz et al., 2015), although Rynkiewicz et al. (2015) reported a higher infection rate (97.8%). In this study, B. grahamii represented 33.9% and 23.9% of bacterial sequences in M. natalensis and M. coucha. Previously detected in Gerbilliscus leucogaster in South Africa and Rattus norvegicus in Nigeria (Pretorius et al., 2004; Kamani et al., 2013), B. grahamii is associated with retinal occlusions, cat scratch disease and neuroretinitis in humans (Kerkhoff et al., 1999; Serratrice et al., 2003; Oksi et al., 2013). The co-infection with other Bartonella spp. highlights the need for further studies on Bartonella’s pathogenicity and transmission dynamics, given its public health implications. The presence of zoonotic B. grahamii in local rodents underscores the potential health risks to the community.
Rodents in peri-urban and communal rangelands had higher Bartonella burdens compared to those in protected areas, indicating a correlation between Bartonella spp. prevalence and human proximity.
Bartonella mastomydis constituted a significant portion of bacterial sequences in M. coucha (35.4%) and M. natalensis (14.9%). The organism has previously been detected in R. tanezumi flavipectus in China (Li et al., 2004). Its presence in South African rodents coupled with the molecular confirmation of invasive R. tanezumi in South Africa (Bastos et al., 2011) suggests spillover of infection between invasive and indigenous species (Hatyoka et al., 2019a).
Analysis of Bartonella gltA gene sequences confirmed B. mastomydis in M. natalensis and M. coucha across habitats. This strain has been found in other South African murids including Aethomys ineptus, Micaelamys natalensis and R. rattus (Mostert, 2010; Brettschneider et al., 2012; Hatyoka et al., 2019a). These studies pre-date the formal description of this species, all noting the close relationship of these strains to Bartonella elizabethae, a zoonotic species, thus 16S rRNA sequencing may misclassify B. mastomydis as B. elizabethae (Kosoy et al., 2018). These results represent the first confirmation of this organism in Mastomys in South Africa. Phylogeny of the gltA gene revealed a new Bartonella sp. in two M. natalensis specimens from Gottenburg and Manyeleti. Another taxon closely related to a Bartonella sp. AN-nh2 previously detected in Micaelamys namaquensis from the Free state, South Africa (Pretorius et al., 2004) was detected in R31 (M. natalensis) from Gottenburg.
Bartonella henselae, known for causing endocarditis and occult infections in immunocompromised humans (Hadfield et al., 1993; Breitschwerdt et al., 2007), was detected in R30 (Gottenburg). Bartonella henselae has been previously detected in cats and humans in South Africa (Kelly, 1996; Frean et al., 2002; Trataris et al., 2012). Its prior detection in two AFI patients in Bushbuckridge-East (Simpson et al., 2018) and now in Mastomys spp. from the same area indicates its potential role in human infections.
Our finding that Mastomys spp. across habitat areas were concurrently infected with multiple Bartonella spp. aligns with prior research showing a variety of Bartonella spp. in Chinese and Spanish rodents (Ying et al., 2002) (Márquez et al., 2008). Similarly, three distinct Bartonella lineages were detected in Rhabdomys pumilio (Western Cape) and A. ineptus (Gauteng) (Hatyoka et al., 2019a, 2019).
As arthropod vectors transmit Bartonella spp. (Breitschwerdt and Kordick, 2000), we examined the ectoparasites from the rodents, recovering 439 ectoparasites. Regulations of the South African Department of Agriculture, Land Reform and Rural Development (DALRRD) [which stated that whole body rodent carcasses must first be frozen at -80°C and stored at Hans Hoheisen Research Centre, Mpumalanga South Africa before transport on dry ice to a BSL3 provincial laboratory at Stellenbosch, South Africa where ectoparasites could be removed] prevented immediate screening of the ectoparasites for Bartonella spp. or any other pathogens at the time of the rodent capture. Nevertheless, the findings of fleas, ticks and mites is consistent with vectored transmission. Results of ectoparasite screening studies are currently ongoing and will be reported at a later time.
Overall, 17% of bacterial sequences in rodent blood corresponded to Pseudomonas spp., primarily from R21 and R159, from Manyeleti, with the organism making 91.6% and 81% of the total sequences obtained from the two samples. Pseudomonas spp. are opportunistic pathogens causing infection in immunocompromised rodents (Baker, 1998), yet their presence could stem from contamination, as they are known contaminants in 16S rRNA gene sequencing (Salter et al., 2014). DNA extraction kits and other laboratory reagents have also been implicated as sources of bacterial DNA contamination in microbiome studies (Salter et al., 2014). However, physical signs like tail growths and foot deformities associated with bumblefoot (ulcerative pododermatitis) observed in some of rodents in this study suggest valid Pseudomonas infections (Blair, 2013) (Supplementary Figure S5).
Ochrobactrum spp. made up 7.2% of sequences from rodent blood, in M. natalensis (R29, R30, R31) from Gottenburg and Steatomys (R53) from Manyeleti. In R53, Ochrobactrum dominated comprising 85% of the total sequences. Recognized as opportunistic nosocomial pathogens (Hagiya et al., 2013), Ochrobactrum can present clinical symptoms that mimic the more virulent Brucella spp. Their close phylogenetic relationship has led to discussions on unifying Ochrobactrum and Brucella, with dissenting views (Hördt et al., 2020; Oren and Garrity, 2020). The CDC has recently unified these taxa (CDC, 2022) and the American Society for Microbiology Clinical and Public Health Microbiology Committee suggested that clinical laboratories report the affected organisms as Brucella (Ochrobactrum) species to differentiate them from the archetypal Brucella agent (She et al., 2023). Recently, 78 scientists highlighted significant flaws in the proposed nomenclature, citing insufficient phylogenetic analysis and exclusion of expert opinion, which could lead to serious risks for personnel handling zoonotic Brucella, particularly in poorer nations (Moreno et al., 2023). Therefore, based on the current knowledge on both organisms, we agree that Ochrobactrum and Brucella spp. be maintained as separate genera and have treated them as such.
Anaplasma centrale and A. marginale were detected in R20 (M. coucha) from Gottenburg and A. phagocytophilum in R98 (M. natalensis) from Hlalakahle. Typically, A. marginale and A. centrale infect cattle and other wild ruminant species (Aubry and Geale, 2011; Hosseini-Vasoukolaei et al., 2014; Wu et al., 2015; Khumalo et al., 2016, 2018). The detection of A. marginale and A. centrale in wild rodents was surprising as these species are thought to only infect ruminants. Because the number of sequences obtained was low, and sequence coverage was high (Rhoads and Au, 2015), their detection could be due to salivary secretions from infected ticks rather than successful establishment of infection. However, rodents in this study had a low tick burden and the Anaplasma positive rodents had no ticks on them at the time of capture. Anaplasma phagocytophilum detection in rodents, dogs and cattle from the study area has been previously reported (Kolo et al., 2020).
An Ehrlichia sp. was identified in R75 from Hlalakahle and R172 from Gottenburg (M. natalensis) with 99% identity to Ehrlichia sp. Ehf669 (AY309969) detected from Haemaphysalis ticks collected from dogs in Japan (Inokuma et al., 2004) and 98% identity to E. chaffeensis (NR_074500). Ehrlichia chaffeensis causes human monocytic ehrlichiosis (HME), a tick-borne zoonosis in the US transmitted by Amblyomma americanum ticks (Paddock and Childs, 2003). Previously, antibodies against E. chaffeensis have been detected in dogs in the Free State (Pretorius and Kelly, 1998). This study reports the first finding of an E. chaffeensis-like sequence in South African rodents.
Coxiella burnetii, the causative agent of Q fever, was detected in R12 (M. coucha) from Tlhavekisa marking its first detection from a wild rodent in South Africa. Q fever, manifesting as an acute febrile illness and chronic endocarditis in humans, and linked to livestock abortion (Vanderburg et al., 2014), is transmitted by ticks, aerosols or consumption of contaminated animal products (Maurin and Raoult, 1999). In South Africa, C. burnetii antibodies have been detected in cattle (Gummow et al., 1987), and wild dogs in the Kruger Park (Van Heerden et al., 1995).
We did not detect any Rickettsia spp. in rodent blood in this study contrasting with Essbauer et al. (2018) who detected pathogenic R. conorii, R. massiliae, R. felis and R. helvetica, in ear tissue of rodents sampled across South Africa and Namibia. Rickettsial pathogens are usually found in the dermis, vascular endothelium and spleen (Hawley et al., 2007; Bayliss et al., 2009) which may explain the absence of detection in blood samples. Essbauer et al. (2018) did not report any Rickettsia in M. natalensis, but found a 9% infection rate in M. coucha, which were identified as “Candidatus Rickettsia africaustralis”.
The detection of Anaplasma, Ehrlichia, B. henselae, and Coxiella in Mastomys spp. in this study was minimal, possibly lacking statistical significance. However, their zoonotic and veterinary significance warranted emphasis.
PCA analysis showed that rodents from Tlhavekisa and Hlalakahle shared similar blood microbiome profiles as opposed to rodents from Tlhavekisa and Gottenburg rodents which had distinct profiles. This is despite Gottenburg being closer to Tlhavekisa than Hlalakahle (7.7 km vs 8.1 km). The blood microbiome of Manyeleti rodents was also distinct, with Pseudomonas and Ochrobactrum spp. dominance. This finding supports studies by Gavish et al. (2014) and Gall et al. (2017) that suggested factors like geography, host diversity, and human interaction might influence bacterial diversity in hosts and vectors. PCA also revealed an association of positive correlations between Bartonella spp. and unclassified Rhizobiales and between Ochrobactrum spp., unclassified Brucellaceae, and B. henselae. Overall, the small sample size meant individual variations significantly influenced the results.
Our results suggest that the rodent blood microbiome is relatively species-sparse. Although the mean sequencing depth was low, the rarefaction curves suggest that the species richness in all samples was adequately captured, and further sequencing would not significantly increase the number of observed species. It is possible that the observed pattern reflects some degree of bias towards dominant species as PCR-based amplification and sequencing methods often favor highly abundant sequences, which may overshadow less abundant or rare species. Under-sampling rare species is a known limitation of low sequencing depths, and future studies could address this by increasing the sequencing depth to capture rare OTUs.
The study identified 17 OTUs across ten species and six genera, with Bartonella grahamii and B. mastomydis dominating the dataset. Approximately 90.0% of the total reads could be assigned to valid taxa, while reads classified as “Rare” accounted for 4.7% of the total reads. This taxonomic distribution indicates that the majority of the reads were assigned to non-rare taxa, suggesting a skew toward dominant species in the microbiome. These findings correspond with previous studies that show rodent blood microbiomes are often dominated by a few bacterial species due to selective pressures and niche specificity in the bloodstream environment (Gavish et al., 2014; Rynkiewicz et al., 2015). Another study found that the rodent blood microbiome is influenced by factors such as host immunity and interspecific bacterial interactions that favors the dominance of specific bacterial species (Cohen et al., 2015). This species-sparse nature of the rodent blood microbiome may explain why a mean sequencing depth of 2,602 reads was sufficient to capture the majority of OTUs present.
In conclusion, this study provides foundational data on bacterial diversity in the blood of indigenous murid rodents and highlights Mastomys spp. as key reservoirs of bartonellae. It reports the first confirmation of B. mastomydis detection in two cryptic Mastomys species in South Africa and documents the detection of important zoonotic pathogens including Ehrlichia spp., and C. burnetii.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statementThe animal study was approved by the University of Pretoria’s Faculty of Veterinary Science animal ethics committee (V105-15). Research permissions for trapping and transporting rodents were granted by the South Africa Department of Agriculture, Land Reform and Rural Development (DALRRD) (12/11/1/1, 12/11/1/1/6), and the Mpumalanga Tourism and Parks Agency (MTPA, B1/290/2016), in accordance with the Animal Diseases Act (Section 20, 1984). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributionsAK: Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. KB: Conceptualization, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – review & editing, Visualization. NC: Conceptualization, Formal Analysis, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. AB: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Software, Writing – review & editing, Resources, Validation, Visualization. SM: Methodology, Resources, Validation, Visualization, Writing – review & editing, Project administration, Investigation. CG: Investigation, Methodology, Software, Writing – review & editing. JW: Methodology, Project administration, Resources, Writing – review & editing. LN: Investigation, Methodology, Project administration, Writing – review & editing, Resources. MO: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. We thank the South African National Research Foundation (NRF) for grants 92739 and 110448, the Foundational Biodiversity Information Program (FBIP) small grant FBIS170330225236, and NIH NIAID R01AI136832 awarded to MO. The Centers for Disease Control and Prevention (CDC) Cooperative Agreement (Co-Ag) 5 NU2GGH001874-02-00 for support awarded to AB and gratefully acknowledge NRF for facility support awarded to the University of Pretoria’s Sanger sequencing facility (grant No: UID78566).
AcknowledgmentsWe acknowledge Anja Le Grange and Liezl Retief (University of Pretoria, Pretoria, South Africa) for assistance with rodent and Bartonella typing and Ivan Horak from the University of Pretoria, South Africa for verifying the tick identification. We also thank the staff of the Hans Hoheisen Wildlife Research Station for logistical support, environmental monitors for assistance in the Bushbuckridge-East community, as well as Charles Byaruhanga, Greg Simpson, Zamatungwa Khumalo, Luther van der Mescht, Götz Froeschke and Conrad Matthee who assisted with rodent sample collection. We thank Estelle Mayhew for production of the map.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimerThe content is solely the responsibility of the authors and does not necessarily represent the official views of the funding bodies.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1520086/full#supplementary-material
ReferencesBastos, A. D. (2007). Bartonella incidence and diversity in endemic South African murid rodents occurring commensally with humans. S. Afri. Soc Vet. Epidemiol. Prev. Med., 78–83.
Bastos, A. D., Chimimba, C. T., Von Maltitz, E., Kirsten, F., Belmain, S. R. (2005). Identification of rodent species that play a role in disease transmission to humans in South Africa. Proc. S. Afr. Soc Vet. Epidemiol. Prev. Med., 78–83. doi: 10.13140/2.1.2629.4563
Crossref Full Text | Google Scholar
Bastos, A. D., Nair, D., Taylor, P. J., Brettschneider, H., Kirsten, F., Mostert, E., et al. (2011). Genetic monitoring detects an overlooked cryptic species and reveals the diversity and distribution of three invasive Rattus congeners in South Africa. BMC Genet. 12, 26–26. doi: 10.1186/1471-2156-12-26
PubMed Abstract | Crossref Full Text | Google Scholar
Bayliss, D. B., Morris, A. K., Horta, M. C., Labruna, M. B., Radecki, S. V., Hawley, J. R., et al. (2009). Prevalence of Rickettsia species antibodies and Rickettsia species DNA in the blood of cats with and without fever. J. Feline Med. Surg. 11, 266–270. doi: 10.1016/j.jfms.2008.06.007
PubMed Abstract | Crossref Full Text | Google Scholar
Berrian, A. M., van Rooyen, J., Martínez-López, B., Knobel, D., Simpson, G. J. G., Wilkes, M. S., et al. (2016). One Health profile of a community at the wildlife-domestic animal interface, Mpumalanga, South Africa. Prev. Vet. Med. 130, 119–128. doi: 10.1016/j.prevetmed.2016.06.007
PubMed Abstract | Crossref Full Text | Google Scholar
Blair, J. (2013). Bumblefoot: a comparison of clinical presentation and treatment of pododermatitis in rabbits, rodents, and birds. Vet. Clin. North Am. Exot. Anim. Pract. 16, 715–735. doi: 10.1016/j.cvex.2013.05.002
PubMed Abstract | Crossref Full Text | Google Scholar
Bonnet, S., Michelet, L., Moutailler, S., Cheval, J., Hebert, C., Vayssier-Taussat, M., et al. (2014). Identification of parasitic communities within European ticks Using next-generation sequencing. PloS Negl. Trop. Dis. 8, e2753. doi: 10.1371/journal.pntd.0002753
PubMed Abstract | Crossref Full Text | Google Scholar
Bonwitt, J., Sáez, A. M., Lamin, J., Ansumana, R., Dawson, M., Buanie, J., et al. (2017). At home with Mastomys and Rattus: human-rodent interactions and potential for primary transmission of Lassa Virus in domestic spaces. Am. J. Trop. Med. Hyg. 96, 935–943. doi: 10.4269/ajtmh.16-0675
PubMed Abstract | Crossref Full Text | Google Scholar
Breitschwerdt, E. B., Kordick, D. L. (2000). Bartonella infection in animals: carriership, reservoir potential, pathogenicity, and zoonotic potential for human infection. Clin. Microbiol. Rev. 13, 428–438. doi: 10.1128/CMR.13.3.428
PubMed Abstract | Crossref Full Text | Google Scholar
Breitschwerdt, E. B., Maggi, R. G., Duncan, A. W., Nicholson, W. L., Hegarty, B. C., Woods, C. W. (2007). Bartonella species in blood of immunocompetent persons with animal and arthropod contact. Emerg. Infect. Dis. 13, 938–941. doi: 10.3201/eid1306.061337
PubMed Abstract | Crossref Full Text | Google Scholar
Brettschneider, H., Bennett, N. C., Chimimba, C. T., Bastos, A. D. (2012). Bartonellae of the Namaqua rock mouse, Micaelamys namaquensis (Rodentia: Muridae) from South Africa. Vet. Microbiol. 157, 132–136. doi: 10.1016/j.vetmic.2011.12.006
PubMed Abstract | Crossref Full Text | Google Scholar
Budachetri, K., Browning, R. E., Adamson, S. W., Dowd, S. E., Chao, C. C., Ching, W. M., et al. (2014). An insight into the microbiome of the Amblyomma maculatum (Acari: Ixodidae). J. Med. Entomol. 51, 119–129. doi: 10.1603/ME12223
PubMed Abstract | Crossref Full Text | Google Scholar
Centers for Disease Prevention and Control (2022). “12/19/2022: Lab Update: Reclassification of Ochrobactrum species into the Brucella genus” (C.f.D.C.a. Prevention).
Cohen, C., Toh, E., Munro, D., Dong, Q., Hawlena, H. (2015). Similarities and seasonal variations in bacterial communities from the blood of rodents and from their flea vectors. ISME J. 9, 1662–1676. doi: 10.1038/ismej.2014.255
PubMed Abstract | Crossref Full Text | Google Scholar
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145. doi: 10.1093/nar/gkn879
PubMed Abstract | Crossref Full Text | Google Scholar
Davis, D. H. (1964). Ecology of wild rodent plague. Reprinted from ecological studies in southern africa. Monogr. Biol. 14, 301–314. doi: 10.5555/19662901642
留言 (0)