
記住我


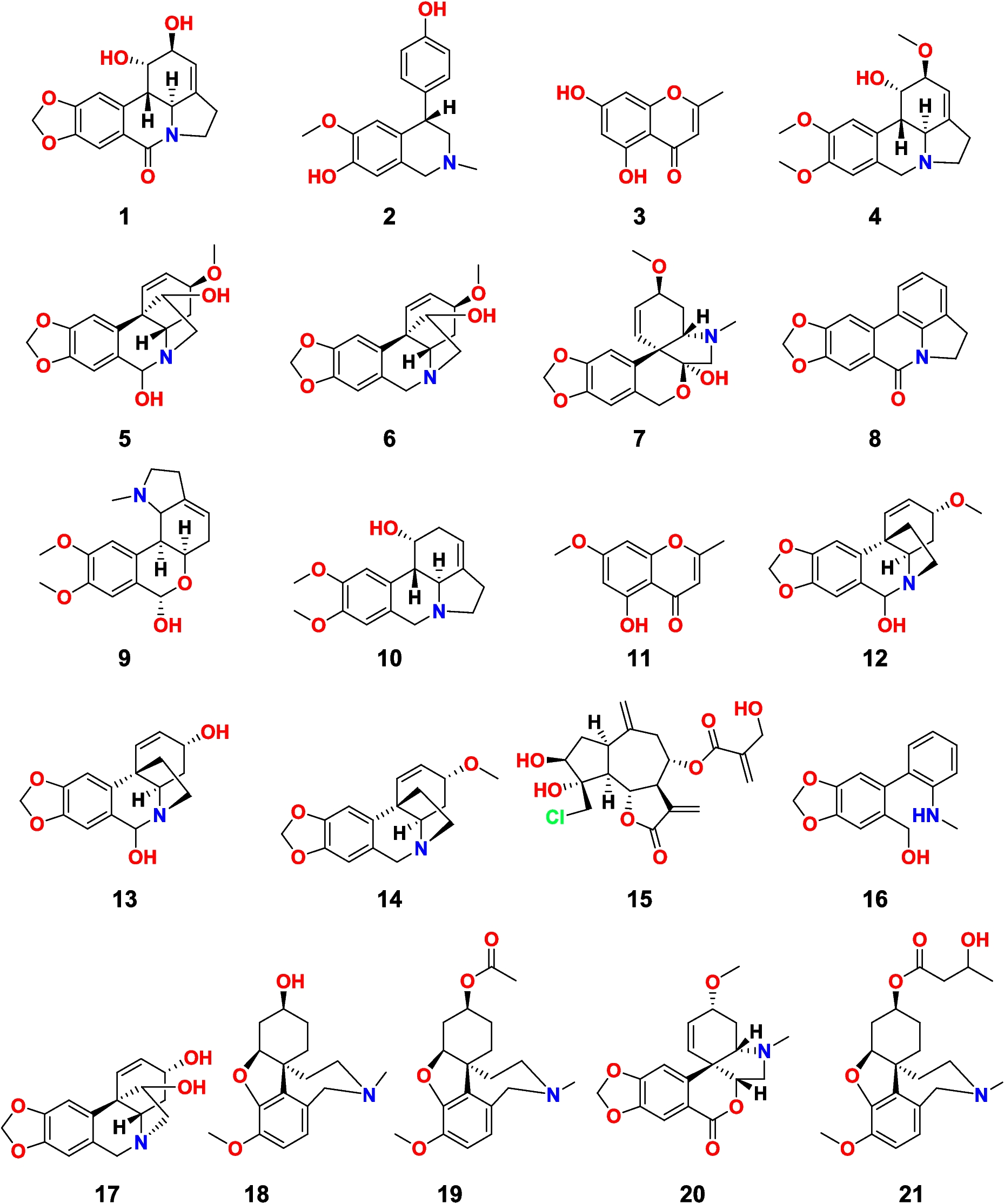
The disk diffusion soft agar colony formation assay could be used as a powerful tool in evaluating the efficacy of different drugs in mitigating the proliferation of cancer cells. This assay determines a zone of inhibition for a sample diffusing from a disk placed at a petri dish containing tumor cells embedded in a soft agar matrix. A decrease in colony formation with respect to an increase in drug concentration indicates effective cytotoxicity of the drug in vitro. It has an advantage over many assays as the semi-solid matrix permits a favorable environment for the anchorage growth of cancer cells which cannot be exhibited by normal cells as well as it mimics the in vivo 3D cellular environment (Horibata, 2015). In this study, 21 compounds isolated from C. bulbispermum, P. maritimem, H. vittatum, and C. scoparia were tested for their tumor growth inhibitory activities to NSCLC cells (H-125).
The zone assay was designed to look for selectivity between the human solid tumor, H125, and either the leukemia (CEM) or the normal cell (CFU-GM). It can also provide a measure of potency. Table 1 shows the initial concentration of the stock solutions of the compounds (column 2), the amount used from these stock solutions in the assay (column 3), and the zone of inhibition against H125, CEM, and CFU-GM (columns 4, 6, 7, respectively). A cutoff of 200 units/µg was used in this assay. Potency is defined as the ratio of the H125 zone and the amount of compound added to the filter disk (columns 6 and 7, respectively). Table 1 displays the potency of 1–21 in descending order with three distinct groups in this table. As shown in Table 1, group A comprises nine compounds (17, 1, 5, 6, 16, 10, 4, 11 and 15) that were considered as active. Crinamine (17) was the most potent one among this group followed by lycorine (1), hemanthidine (5), and haemanthamine (6), and Group B comprises a set of four compounds (19, 12, 20, and 21) that had H125 zones of between 100 and 200 zone units. In our studies, we usually call these as moderately active, since a cutoff of above 200 zone units was set for activity. Finally, group C with eight compounds (2, 3, 7, 8, 9, 13, 14, and 18) is considered inactive because there are no zones of inhibition observed. Only one compound, pluviine (10), demonstrated any solid tumor selectivity, H125ΔCFU = 275 units.
Table 1 Disk diffusion assay results of compounds 1–21The synergistic effect is such an important criterion especially when considering candidates for cancer treatment (Bayat Mokhtari 2017). Therefore, an in vitro assay was also carried out to investigate the synergistic effect of crinamine (17) (being the most in vitro active compound among the tested candidates) and ismine (16) (the top hit as revealed from subsequent in silico studies). Surprisingly, this combination exhibited a synergistic effect on H-125 cells (Table 1). Where the H125 zones exceeded 1000 zone units for the combination, exceeding their individual additive zone units, 500 and 350 zone units for crinamine and ismine, respectively. Therefore, this approach promotes this combination for potential treatment of NSCLC.
Network pharmacology analysisTarget fishing and construction of networksTo examine the profound pharmacological mechanisms of the most active isolates in treating NSCLC, interactions with the related targets and identification of involved pathways were investigated using “Network Pharmacology” analysis (Shi et al. 2019).
The top 50 potential targets having the highest interaction combined scores for each of the active compounds were analyzed with the top 200 NSCLC-related targets using Venny 2.1.0 aiming to identify the possible common genes (Fig. 2, Table 2). Common genes varied from one compound to another, where acetyllycoramine (19) shared the highest number of overlapping genes with NSCLC (5 genes), followed by ismine (16), pluviine (10), and 5-hydroxy-7-methoxy-2-methylchromone (11) (4 genes), while crinamine (17) showed interactions with only one NSCLC-related gene.
Fig. 2
Venn diagrams illustrating the number of NSCLC-related genes shared by each of the top-scored compounds
Table 2 Information on potential NSCLC-related protein targets shared with the top scored isolated compoundsIt did not escape our notice that the results obtained from network pharmacology analyses deviated slightly from those of the in vitro cytotoxic activity assay. This was probably due to the difference in pharmacokinetic profiles of the different compounds including hydrophobicity, solubility, molecular size, and weight that might significantly affect the penetration power of these compounds through the cancer cells to exert their cytotoxic activity. This prompted us to conduct further in silico pharmacokinetic studies to better understand their properties and provide suggestions for pharmaceutical and structural modifications to achieve enhancement in the compound’s permeability.
Meanwhile, a total of 13 targets, distributed among the compounds, were profoundly related to NSCLC thus, recognized as candidate targets. Inspection of the targeted genes (Fig. 3, Table 2) indicated that the genes AR, EGFR, and ESR-1 targets were the most enriched genes being common to compounds 6, 4, and 3, respectively. This suggested their important role in the entire pathogenesis of NSCLC as well as the synergistic effect of these compounds in treating NSCLC, as they possibly act on some of the same biological processes or pathways. Several studies have documented the relationship between those genes and inflammation. For example, it was shown that male patients diagnosed with lung cancer exhibited greater survival when exposed to androgen pathway manipulation (APM) (Harlos et al. 2015). Also, cell proliferation, migration, invasion, and tumor formation were repressed by the interference of androgen receptor (AR) in NSCLC cell lines (Yeh et al. 2012). Tumor xenograft assay experiments confirmed that NSCLC tumorigenesis could be abolished by stimulating AR and epithelial-mesenchymal transition (EMT) mediation (Zhou et al. 2021). In the same context, drugs possessing dual action on VEGF-EGFR receptors have been approved and established for use in NSCLC (Le et al. 2021). Moreover, targeting FBXL2-Grp94 pathways might serve as a promising therapeutic strategy for tyrosine kinase inhibitors (TKI)-resistant NSCLC (Niu et al. 2021).
Fig. 3
A donut chart showing the number of constituents interacting with each of the determined common genes
The importance of ESR-1 in improving the prognostic outcome of NSCLC was investigated using anti-hormonal drugs (Skjefstad et al. 2016; Enwere et al. 2020). Meanwhile, EGFR and ER were observed to cooperate in early activation of p42/p44 MAP kinase in NSCLC cells; hence, the synergistic use of antiestrogens with growth factor receptor antagonists could lead to the development of novel palliative strategies in NSCLC treatment.
Drawing support from target fishing, the aforementioned lung cancer targets were forwarded to KEGG functional enrichment analyses using the STRING database. KEGG analysis showed that the manifestation and progress of lung cancer were related to 39 NSCLC-related pathways, the most strikingly associated with NSCLC were pathways in cancer, proteoglycans in cancer, central carbon metabolism in cancer, EGFR tyrosine kinase inhibitor resistance, and endocrine resistance (Table 3).
Table 3 KEGG pathway analysis of potential target genes functionsInteractions between the top-scored compounds with the proteins and pathways involved in NSCLC were unveiled via constructing a constituent-target-pathway network (Fig. 4) including 48 nodes (11 compound nodes, 13 target nodes, 37 pathway nodes) and 198 edges. These data revealed the poly-pharmacology and multi-target properties of these compounds.
Fig. 4
Network of compound-target gene-pathway interactions for the top-scored isolates with 13 target proteins and 39 pathways presented by violet, orange, and green heptagon nodes, respectively
Protein–protein interactions were examined using the STRING database and then visualized through P-P network analysis. From this network, strong correlations between the identified potential NSCLC target proteins were spotted, suggesting that they probably regulate the functions of each other (Fig. 5).
Fig. 5
Protein–protein interaction (PPI) network of the putative NSCLC-related targets
Gene ontology enrichment analysisCommon genes were subjected to GO enrichment analysis using the DAVID database to illustrate the underlying mechanism of the top-scored isolates in mitigating NSCLC using biological process (BP), cellular component (CC), and molecular function (MF) terms. For instance, BP describes gene participation from the cellular to systemic level. CC presents the protein location inside/outside the cell, while MF demonstrates the protein functions at the molecular level (Lagunin et al. 2020). It is considered a statistically-based approach intended for the determination of over-expressed groups of proteins that may have an association with a certain disease (Lagunin et al. 2020). According to preliminary results (Fig. 6), a total of 40 BP terms were selected, the most related to NSCLC included positive regulation of protein kinase-B signaling, peptidyl-tyrosine phosphorylation, signal transduction, transmembrane receptor protein tyrosine kinase signaling pathway, and protein autophosphorylation. Concurrently, five key targets were primarily distributed in cellular components such as receptor complex, plasma membrane, basal plasma membrane, perinuclear region of cytoplasm, and cytosol. According to enrichment analysis of molecular functions, the targets were mainly involved in protein tyrosine kinase activity, protein serine/threonine/tyrosine kinase activity, transmembrane receptor protein, tyrosine kinase activity, ATP binding, and protein kinase activity.
Fig. 6
Gene ontology analysis of the putative targets of NSCLC by the DAVID database. BP, MF, and CC terms are represented by orange, green, and blue bars, respectively. The significance of enrichment is indicated by log p-value with bar charts. The blue line represents the number of genes enriched by each term
Pathways functional enrichment analysis (Fig. 7) allowed for the determination of the signaling pathways and functions of identified target genes where 46 KEGG pathways were recognized (p < 0.05). Pathways in cancer were the most enriched with the highest number of observed genes and lowest false discovery rate followed by central carbon metabolism in cancer, EGFR tyrosine kinase inhibitor resistance, and endocrine resistance. Further, NSCLC was observed to be strongly modulated by 12 BIOCARTA pathways, the most positively associated of which were ERBB2 in signal transduction and oncology and CBL-mediated ligand-induced downregulation of EGF receptors.
Fig. 7
Major KEGG (orange) and BIOCARTA (green) pathways clusters generated from the DAVID database. The significance of enrichment is indicated by log p-value with bar charts. Blue lines represent the number of genes enriched by each term
Based on the above network pharmacology analysis results, the four top-scored compounds named acetyllycoramine (19), ismine (16), pluviine (10), and 5-hydroxy,7-methoxy-2-methylchromone (11) were subjected to molecular docking studies aiming to afford deepened overview of their multi-targeted molecular mechanisms.
Molecular docking and dynamics studiesFirstly, we made use of the RMSD value for validation of the docking protocol which was obtained through the use of the ligand co-crystallized with each crystal structure (PDB ID: 2PIW, 1M17, and 6CBZ). As highlighted in Table 4, for the three protein target crystal structures, the RMSD value was less than 1 A, which reflects high docking accuracy (Dawood et al. 2023; Ibrahim et al. 2021).
Table 4 RMSD and XP-G scores of the top four scoring compounds and erlotinib docked into AR, EGFR and ESR-1 active sites (expressed in kcal/mol)To study the molecular interactions between the top four hit constituents and the three most enriched target genes (AR, EGFR, and ESR-1), molecular docking studies were conducted, and their 2D and 3D interaction patterns are shown in Figs. 8, 9, and 10. Among all of the tested ligands analyzed, ismine (16) performed best in multi-regimen inhibition of lung cancer cell growth. It showed efficient binding, with all three receptor proteins having the most negative binding energies among all ligand–receptor interactions, followed by 5-hydroxy-7-methoxy-2-methylchromone (11), pluviine (10), and finally acetyllycoramine (19) (Table 4). Energetically, ismine (16) and AR exhibited the most favored interaction, with a binding energy of − 9.304 kcal/mol. During this interaction, two hydrogen bonds were formed with the amino acids Leu704 and THR877, 16 hydrophobic interactions with backbone amino acid residues in addition to two polar interactions with ASN705 and Leu704 (Fig. 8a and b). Additionally, Fig. 8c represents the 2D interaction diagram of the co-crystallized ligand (dihydrotestosterone).
Fig. 8
a 2D and b 3D ligand interaction diagrams for docking poses of ismine in the active site of AR crystalline structure (PDB ID: 2PIW) together with c 2D interaction diagram of the co-crystallized ligand dihydrotestosterone
Fig. 9
a 2D and b 3D ligand interaction diagrams for docking poses of ismine in the active site of ESR-1 crystalline structure (PDB ID: 6CBZ) together with (c) 2D interaction diagram of the co-crystallized ligand estradiol
Fig. 10
a 2D and b 3D ligand interaction diagrams for docking poses of ismine in the active site of EGFR crystalline structure (PDB ID: 1m17) together with c 2D interaction diagram of the co-crystallized ligand erlotinib
Concerning docking into the ESR-1 pocket, results showed that ismine (16) displayed weaker binding affinity (− 9.104 kcal/mol) where it shared one hydrogen bonding with Leu346, 13 hydrophobic interactions, two polar interactions with THR347 and HIE524, as well as a positive- and negative-charged interactions with ARG394 and GLU353, respectively (Fig. 9a and b). Moreover, Fig. 9c explains the 2D interaction diagram of the co-crystallized ligand, estradiol.
As shown in the 2D interaction diagram of ismine (16) with EGFR (Fig. 10a and b), it extended into the active site via two hydrogen bondings with the amino acids GLN767 and ASP831, ten hydrophobic interactions, three polar interactions with GLN767, THR766, and THR830, and two negative interactions with ASP831 and GLU738 beside another positive interaction with LYS721. In terms of docking scores, ismine (16) displayed the weakest binding affinity, scoring (− 7.529 kcal/mol), when compared to the other receptor proteins. Finally, Fig. 10c shows a 2D interaction diagram of the co-crystallized ligand; erlotinib.
It can also be noted that ismine (16) showed similar interaction patterns to that of the co-crystalized ligands dihydrotestosterone, estradiol, and erlotinib inside the pockets of AR, ESR-1, and EGFR proteins, respectively (Figs. 8c, 9c, and 10c). This similarity in interactions between ismine and the correct amino acid residues of the three protein targets confirmed that ismine was successfully docked into the correct active sites. Importantly, the presence of ismine in the proper pockets of the three enzymes represents a high potential for this ligand to exert its action with the same underlying mechanism of action. Eventually, this triple action of ismine on the three main top targets of NSCLC urged it as a pharmacologically active candidate for this disease.
For the sake of confirmation of the potential activity of ismine, its docking scores and interactions were compared to erlotinib. Erlotinib is an orally administered small molecular inhibitor of lung cancerous cell line. It causes G0/G1 cell cycle arrest and inhibits cancer cell proliferation. In sensitive cells, erlotinib causes tumor cell apoptosis (Piperdi and Perez-Soler 2012). After performing molecular docking analysis of erlotinib on the three targets—AR, ESR-1, and EGFR—it was astonishing to observe that ismine possessed more favorable docking scores with both AR and ESR-1 compared to erlotinib which possessed docking scores of − 3.800 and − 9.060 kcal/mol, respectively. Meanwhile, erlotinib exhibited a slightly better score (− 9.034 kcal/mol) on EGFR (Table 4). These results raised ismine as a promising candidate for the treatment of NSCLC. The 2D interaction diagrams of erlotinib with AR, ESR-1, and EGFR are depicted in (Fig. S2) in the supplementary materials.
While the number of interactions between the amino acid residues and a compound does not necessarily reflect its true pharmacological activity as the type, the strength of interactions and their inter-site distances play a crucial role in stabilizing the ligand-protein complex which in turn affects its binding affinity (Khaerunnisa, 2020). That could explain the lower docking score of ismine (16) when bound to EFGR compared to ESR-1 despite possessing a higher number of interactions.
To validate the docking results and explore the dynamic stability of ismine in complex with AR (PDB ID, 2PIW), ESR-1 (PDB ID, 6CBZ), and EGFR (PDB ID, 1M17), 100-ns molecular dynamics (MD) simulations were conducted. The MD trajectories were analyzed for stability, key interactions, and conformational changes over time (Fig. 11a and b).
Fig. 11
a Protein–ligand RMSD analysis over 100 ns for AR, ESR-1, and EGFR with ismine, showing stability and fluctuations. b Protein Rg analysis indicating compactness during the simulation. c Interaction map of AR-ismine at 100 ns, showing stable key interactions. d Interaction map of ESR-1-ismine at 100 ns, with consistent interactions and enhanced contacts. e Interaction map of EGFR-ismine at 100 ns, displaying maintained interactions with minor flexibility
The MD simulation of the AR-ismine complex demonstrated high stability over 100 ns, with RMSD values stabilizing at ~ 0.2 to 0.3 nm, indicating minimal conformational changes, and the radius of gyration (Rg) values consistently around 1.75–1.8 nm, reflecting a compact structure. Key hydrogen bonds with LEU704 and THR877 observed in the docking pose were preserved throughout the simulation, along with polar interactions with ASN705. Hydrophobic contacts with residues such as PHE876, MET742, and LEU707 remained consistent for time (Fig. 11c). The 100-ns pose showed slight adjustments that optimized hydrogen bonding and hydrophobic interactions, aligning closely with the docking pose and confirming AR as the most stable binding target for ismine (Video S1).
In the ESR-1-ismine complex, RMSD values were slightly higher (~ 0.3 to 0.4 nm), reflecting moderate stability, and the Rg values remained consistent at ~ 1.8 to 1.85 nm, indicating a stable but less compact structure compared to AR (Fig. 11B). Hydrogen bonding with LEU346 was retained, but the bond with GLU353, observed in the docking pose, was intermittently lost. Hydrophobic interactions with residues such as LEU387, LEU391, and MET421 remained stable, while additional contacts with deeper residues like LEU428 emerged over time (Fig. 11d). The 100-ns pose exhibited some deviation from the docking pose, with slight rearrangements enhancing hydrophobic contacts and compensating for the loss of some polar interactions (Video S2).
The EGFR-ismine complex showed the highest RMSD (~ 0.4 to 0.6 nm), indicating lower stability and greater flexibility, with Rg values fluctuating between 1.85 and 1.9 nm, reflecting a more dynamic and less compact structure. While the hydrogen bond with ASN818 was maintained, the bond with SER696 was lost early in the trajectory. Hydrophobic contacts with residues such as ALA719, VAL702, and CYS773 persisted but weakened due to the ligand’s flexibility (Fig. 11e). The 100-ns pose deviated significantly from the docking pose, with reduced hydrogen bonding and weaker hydrophobic interactions, confirming the relatively lower stability of ismine in the EGFR binding pocket compared to AR and ESR-1 (Video S3).
The MMPBSA results align with the MD simulation findings (Table S1), highlighting AR as the most stable and favorable binding target for ismine, with a binding energy of − 90.6 kJ/mol, driven by strong van der Waals and electrostatic interactions. The ESR-1 complex showed moderate stability, with a binding energy of −83.6 kJ/mol, reflecting a balance between hydrophobic and polar contributions despite the loss of some hydrogen bonds during the simulation. In contrast, the EGFR complex exhibited the weakest binding energy (− 39.5 kJ/mol), attributed to weaker van der Waals forces and higher polar solvation energy, consistent with its higher RMSD, fluctuating Rg, and dynamic behavior. These results confirm AR as the most suitable target for ismine, followed by ESR-1, with EGFR showing limited stability.
In silico ADMET studyQikProp was used to determine the ADMET properties of the top four hit compounds (10, 11, 16, 19). It is a useful tool that can provide information regarding the drug-likeness of the compound, its oral absorption, permeability to barriers, metabolic reactions, and toxicity. This valuable information can give a guide to possible chemical modifications to improve its activity (Divyashri et al. 2021).
The main objective of this study was to determine the drug-likeness properties of the tested compounds. This was attained by checking whether they obeyed Lipinski’s rule of five which includes some descriptors with specific ranges named Mol_MW < 500, Qplogpo/w < 5, Donorhb ≤ 5, and Accpthb ≤ 10 (Saini et al. 2022). Analyzing Lipinski’s rule of five for the four compounds (Table 5) showed that all of them exhibited no violations for all these descriptors implying their high drug-likeness potentials. Oral absorption is assessed by some parameters such as the predicted aqueous solubility (QPlogS, − 6.0 to 0.5), the predicted percentage of human oral absorption (25–80%), and obeying Jorgensen’s rule of three. A compound complying with all or some of the rules (QPlogS > − 5.7, Caco2 > 22 nm/s, and # primary metabolites < 7) is considered to be orally available with probable high therapeutic activity as it possessed good aqueous solubility (represented by QPlogS) and satisfying hydrophobicity (represented by Caco2) so that it can easily penetrate the gut walls (James et al. 2023). As depicted in Table 5, all the tested compounds exhibited satisfying values for QPlogS, great percentages of human oral absorption, and successfully obeyed the rule except for pluviine (10) which exceeded the maximum allowed number of metabolic reactions (#metb = 8), thus alerting for the need to apply some chemical and structural modifications to enhance its oral bioavailability.
Table 5 In silico ADMET profiles of the top four hit compoundsAs well known, the binding of the drugs to plasma proteins reduces the concentration of the drug in the bloodstream, thus, abolishing its efficiency. The drug binding to the plasma protein albumin can be predicted by the parameter (QPlogKhsa) with a recommended range of − 1.5 to 1.5 (Sharma et al. 2024). All compounds lay in the recommended range (Table 5), suggesting their free blood circulation and their good availability to the target site.
The human Ether-a-go-go-related gene (hERG) encodes the pore-forming subunit of the rapidly activating delayed rectifier potassium channel (IKr), which is important for cardiac electrical activity and heart beating. Dysfunction of hERG causes long QT syndrome and sudden death, which occur in patients with cardiac ischemia (Omoboyowa 2022). The predicted IC50 of drugs causing channel inhibition by 50% can be estimated using QPlogHERG. All the phytochemicals showed predicted IC50 value above the critical recommended value (− 5) indicating their low cardiotoxic effects.
留言 (0)