
記住我
The term “disorders/differences of sex development” (DSD) indicates a group of congenital conditions with atypical development of chromosomal, gonadal, or anatomical sex. The 46, XY DSD are characterized by ambiguous external genitalia with a 46, XY karyotype (1). Advances in genomics, particularly NGS, have helped identify multiple genes associated with 46, XY DSD, including NR5A1. Steroidogenic factor 1 (SF-1), encoded by the NR5A1 gene, is a transcription factor crucial for adrenal and gonadal organogenesis (2, 3). NR5A1 mutations have been detected in about 10%–20% of 46, XY DSD cases as the major causes of gonadal dysgenesis with a wide range of clinical phenotypes (4, 5). We present the case of a patient with ambiguous genitalia at birth who was investigated for DSD genetic panel conditions, emphasizing the importance of genetic analysis in understanding and managing DSD cases. This individualized approach is crucial for ensuring the well-being of patients and their families, both physically and mentally (6).
Case description Patient informationA 4-month-old male was referred to our Institution with severe genital malformation.
Clinical findingsGenitalia examination revealed a micropenis with complete bifid scrotum and perineal hypospadias (Figure 1). The Glans-Urethral Meatus-Shaft (GMS) hypospadias score (7) was G2M4S4 with complete penoscrotal transposition. Both gonads were palpable at the emergency of the labioscrotal fold and 49 stretched penile length was 2.6 cm.
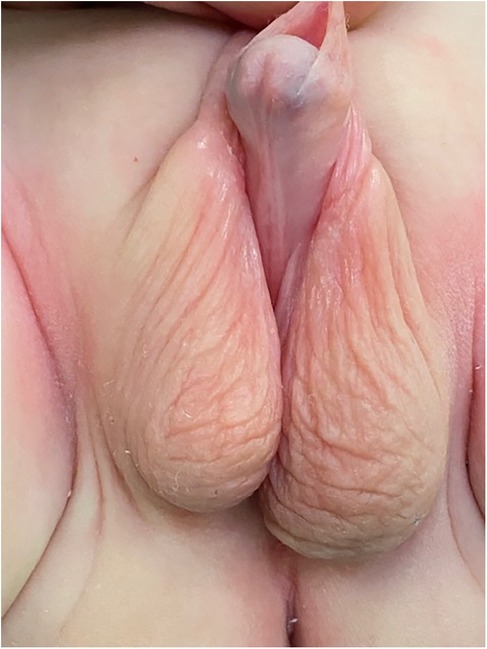
Figure 1. Pre-operative appearance of the external genitalia, complete penoscrotal trasposition.
Diagnostic approachThe cytogenetics results revealed that the patient had a 46, XY karyotype and laboratory workup showed normal hormone levels. Testicular and scrotal ultrasound examination showed both testes in the scrotal sac, symmetrical and equal in size. We collected blood samples from our patient and analyzed his DNA using a DSD 19-gene panel, NGS identified a heterozygous missense variant in NR5A1 gene, classified as a variant of uncertain significance (VUS).
Surgical procedureMultidisciplinary counseling led to surgical intervention, involving a three-stage hypospadias repair (8). In the first stage, at 11 months, a perimeatal incision followed by complete degloving of penile skin up to the level of the penoscrotal junction was made. Artificial erection showed a curvature greater than 60° that was corrected through multiple transverse incisions (corporotomies) of the tunica albuginea in the penile ventral aspect, thus obtaining a 1.2 cm lengthening of the shaft (Figure 2). The corporotomies were covered by a dartos flap from the adjacent shaft skin on one side. Subsequently, a prepuctioplasty was performed: the ventral skin was lengthened with two incisions, the penoscrotal junction was fixed, and the foreskin edges were approximated in two layers ventrally. In the second stage, performed six months after the first step, a dorsal inner preputial flap of 3 × 1.5 cm was harvested and placed ventrally between the proximal meatus and tip of the glans. In the last surgical step, six months after the second surgical step, a U-shaped incision demarcating the neourethra and extending from the original meatus to the distal glans was accomplished. According to the Duplay technique, a three-layer urethral closure was performed. Glanuloplasty and skin cover completed the repair by leaving a wide meatus at the tip of the glans. The urinary diversion was maintained by a urethral catheter and suprapubic cystostomy for 14 days.
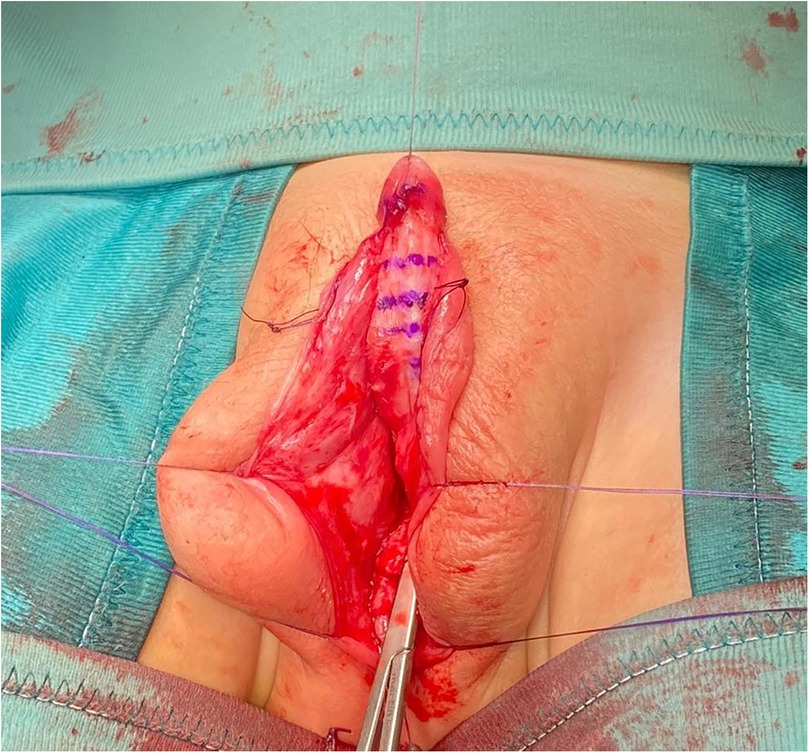
Figure 2. Intra-operative view after the ventral corporotomies.
Postoperative outcomeAn 18-month follow-up revealed an uneventful recovery, with a satisfactory genital appearance (Figure 3).
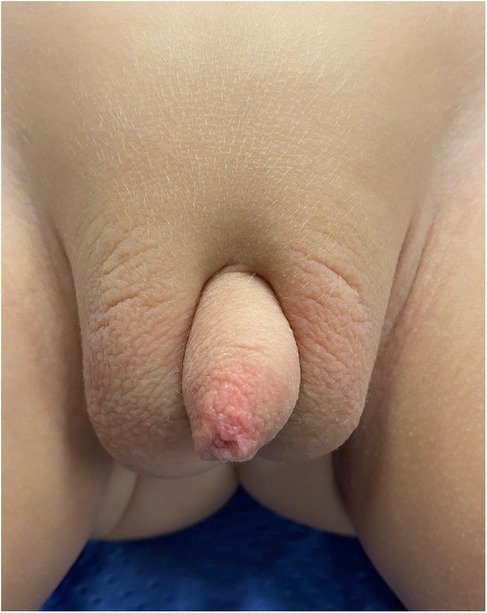
Figure 3. Post-operative appearance after 18-months follow-up.
DiscussionDSDs include a spectrum of phenotypes defined by congenital conditions in which chromosomal, gonadal, or anatomical sex is atypical. Hypospadias is the most frequent form of DSD due to a disruption of the development of the ventral face of the penis in which, the most evident feature is the urethral meatus in an ectopic position. Most often, hypospadias is relatively mild and a genetic cause is not searched or identified (9). However, severe forms, such as posterior and perineal hypospadias with micropenis and significant chordee, represent some of the most severe 46,XY DSDs and may result in ambiguous genitalia at birth. DSDs with a genital appearance that is sufficiently atypical to prompt evaluation occur in approximately 1 in 1,000–4,500 live births (10). The clinical and psychosocial impact of hypospadias is significant, requiring surgical correction typically around one year of age. In severe cases, hypospadias may compromise sexual function, affect quality of life, and impair psychosocial development (11). While most cases are isolated, some are associated with a more complex genetic syndrome involving anomalies of genital development. Moreover, hypospadias may present alongside other abnormalities such as undescended testis, bifid scrotum or inguinal hernia. Overall, less than 10% of cases may be related to genetic defects (12). Familial clustering is present in about 10% of patients with hypospadias. The inheritability is estimated between 57% and 77% of affected patients. Severe hypospadias are generally seen as isolated cases, while milder variants are more often familial (13). The etiology of hypospadias is multifactorial (14, 15). Several genes have been founded to link with hypospadias such as DGKK, ESRs, HOX, ATF3 and VAMP. The precise role of androgen and estrogen signaling that interact in a temporal and tissue-specific manner during genital tubercle development may explain the wide range of meatal/urethral defects. Significant alterations in DNA methylation of sex hormone receptor genes (ESR1 and AR) and fibroblast growth factor (FGFR2 FGF8) may correlate with abnormal expression of these genes in patients with hypospadias. These findings suggest a potential role for epigenetic modifications in hypospadias etiology that, potentially, can be transmitted to future generations. In our case, the patient presented with severe genital malformation, characterized by perineal hypospadias, micropenis, bifid scrotum, and penoscrotal transposition, linked to a heterozygous missense variant in the NR5A1 gene. Although the variant was classified as a variant of uncertain significance (VUS), its identification necessitated a multidisciplinary approach that guided surgical interventions and emphasized the need for long-term follow-up. Mutations in NR5A1 are associated with a broad spectrum of phenotypes, including testicular dysgenesis, progressive endocrine dysfunction and reproductive challenges (3). These factors may also contribute to the risk of gender dysphoria in later life, particularly in cases of severe genital ambiguity. The interplay between physical characteristics, hormonal influences, and psychosocial factors requires ongoing monitoring and psychosocial support. Moreover, the development of gender identity in individuals with NR5A1 mutations may progress over time, necessitating adaptability in management approaches. Comprehensive follow up, including endocrine, psychological, and surgical assessments, is crucial to ensure holistic care and address any emerging concerns, such as declining testosterone production, fertility preservation, or the risk of germ cell tumors (3). Genetic counseling remains essential for the patient and their family, as NR5A1 variants can have implications for female relatives, including the potential for primary ovarian insufficiency (16). The limitations of the studies trying to correlate hypospadias and genetics are that the etiology of hypospadias is obviously multifactorial. These studies are often conducted on a limited number of cases and on a single ethnic group or race. In addition, the various cases of hypospadias are not the same, and the results of the studies are often discordant. To overcome these problems, multicenter studies are needed.
ConclusionThis case underscores the significance of genetic testing in severe hypospadias, highlighting its contribution to multidisciplinary management and long-term monitoring. The identification of an NR5A1 variant, classified as a variant of uncertain significance (VUS), informed surgical decisions and heightened awareness of possible endocrine complications.
Comprehensive care must emphasize endocrine monitoring, psychosocial support, and fertility preservation. Genetic counseling is essential for addressing familial implications and customizing patient-specific strategies. Additional research is required to delineate more precise correlations between genetic findings and clinical outcomes in severe DSD cases.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statementWritten informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributionsGR: Data curation, Writing – original draft. GR: Data curation, Methodology, Writing – original draft. LS: Conceptualization, Writing – original draft. MM: Data curation, Formal Analysis, Writing – review & editing. OA: Writing – review & editing. MS: Supervision, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported also by the Italian Ministry of Health with “Current Research funds”.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. EAU. EAU Guidelines. Edn. Presented at the EAU Annual Congress Paris 2024. ISBN 978-94-92671-23-3. Arnhem, The Netherlands: EAU Guidelines Office (2024). Available online at: https://uroweb.org/guidelines (Accessed November 08, 2024).
3. Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab. (2015) 29(4):607–19. doi: 10.1016/j.beem.2015.07.004
PubMed Abstract | Crossref Full Text | Google Scholar
4. Alhamoudi KM, Alghamdi B, Aljomaiah A, Alswailem M, Al-Hindi H, Alzahrani AS. Case report: severe gonadal dysgenesis causing 46,XY disorder of sex development due to a novel NR5A1 variant. Front Genet. (2022) 13:885589. doi: 10.3389/fgene.2022.885589
PubMed Abstract | Crossref Full Text | Google Scholar
6. Zhang D, Wang D, Tong Y, Li M, Meng L, Song Q, et al. A novel c.64G > T (p.G22C) NR5A1 variant in a Chinese adolescent with 46,XY disorders of sex development: a case report. BMC Pediatr. (2023) 23(1):182. doi: 10.1186/s12887-023-03974-7
PubMed Abstract | Crossref Full Text | Google Scholar
7. Merriman LS, Arlen AM, Broecker BH, Smith EA, Kirsch AJ, Elmore JM. The GMS hypospadias score: assessment of inter-observer reliability and correlation with post-operative complications. J Pediatr Urol. (2013) 9(6):707–12. doi: 10.1016/j.jpurol.2013.04.006
PubMed Abstract | Crossref Full Text | Google Scholar
8. Snodgrass W, Bush N. 2-stage STAG vs 3-stage STAC for primary proximal hypospadias repair. J Pediatr Urol. (2024):S1477513124005473. doi: 10.1016/j.jpurol.2024.10.023
Crossref Full Text | Google Scholar
9. Kalfa N, Gaspari L, Ollivier M, Philibert P, Bergougnoux A, Paris F, et al. Molecular genetics of hypospadias and cryptorchidism recent developments. Clin Genet. (2019) 95(1):122–31. doi: 10.1111/cge.13432
PubMed Abstract | Crossref Full Text | Google Scholar
10. Hughes IA, Nihoul-Fékété C, Thomas B, Cohen-Kettenis PT. Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development. Best Pract Res Clin Endocrinol Metab. (2007) 21(3):351–65. doi: 10.1016/j.beem.2007.06.003
PubMed Abstract | Crossref Full Text | Google Scholar
11. Örtqvist L, Fossum M, Andersson M, Nordenström A, Frisén L, Holmdahl G, et al. Sexuality and fertility in men with hypospadias; improved outcome. Andrology. (2017) 5(2):286–93. doi: 10.1111/andr.12309
PubMed Abstract | Crossref Full Text | Google Scholar
12. Carmichael SL, Shaw GM, Lammer EJ. Environmental and genetic contributors to hypospadias: a review of the epidemiologic evidence. Birth Defects Res. (2012) 94(7):499–510. doi: 10.1002/bdra.23021
PubMed Abstract | Crossref Full Text | Google Scholar
13. Nordenvall AS, Frisén L, Nordenström A, Lichtenstein P, Nordenskjöld A. Population based nationwide study of hypospadias in Sweden, 1973 to 2009: incidence and risk factors. J Urol. (2014) 191(3):783–9. doi: 10.1016/j.juro.2013.09.058
PubMed Abstract | Crossref Full Text | Google Scholar
14. Turkyilmaz Z, Emaratpardaz N, Karabulut R, Kaya C, Atan A, Sonmez K. The role of genetics in the etiology of hypospadias. J Pediatr Urol. (2024):S1477513124005102. doi: 10.1016/j.jpurol.2024.09.025
Crossref Full Text | Google Scholar
15. Yıldız S, Inanç I, Zhuri D, Atlı E, Avlan D. The possible role of epigenetics in the etiology of hypospadias. J Pediatr Urol. (2024) 20(5):877–83. doi: 10.1016/j.jpurol.2024.07.001
Crossref Full Text | Google Scholar
16. Allali S, Muller J-B, Brauner R, Lourenço D, Boudjenah R, Karageorgou V, et al. Mutation analysis of NR5A1 encoding steroidogenic factor 1 in 77 patients with 46, XY disorders of sex development (DSD) including hypospadias. PLoS One. (2011) 6(10):e24117. doi: 10.1371/journal.pone.0024117
留言 (0)