
記住我
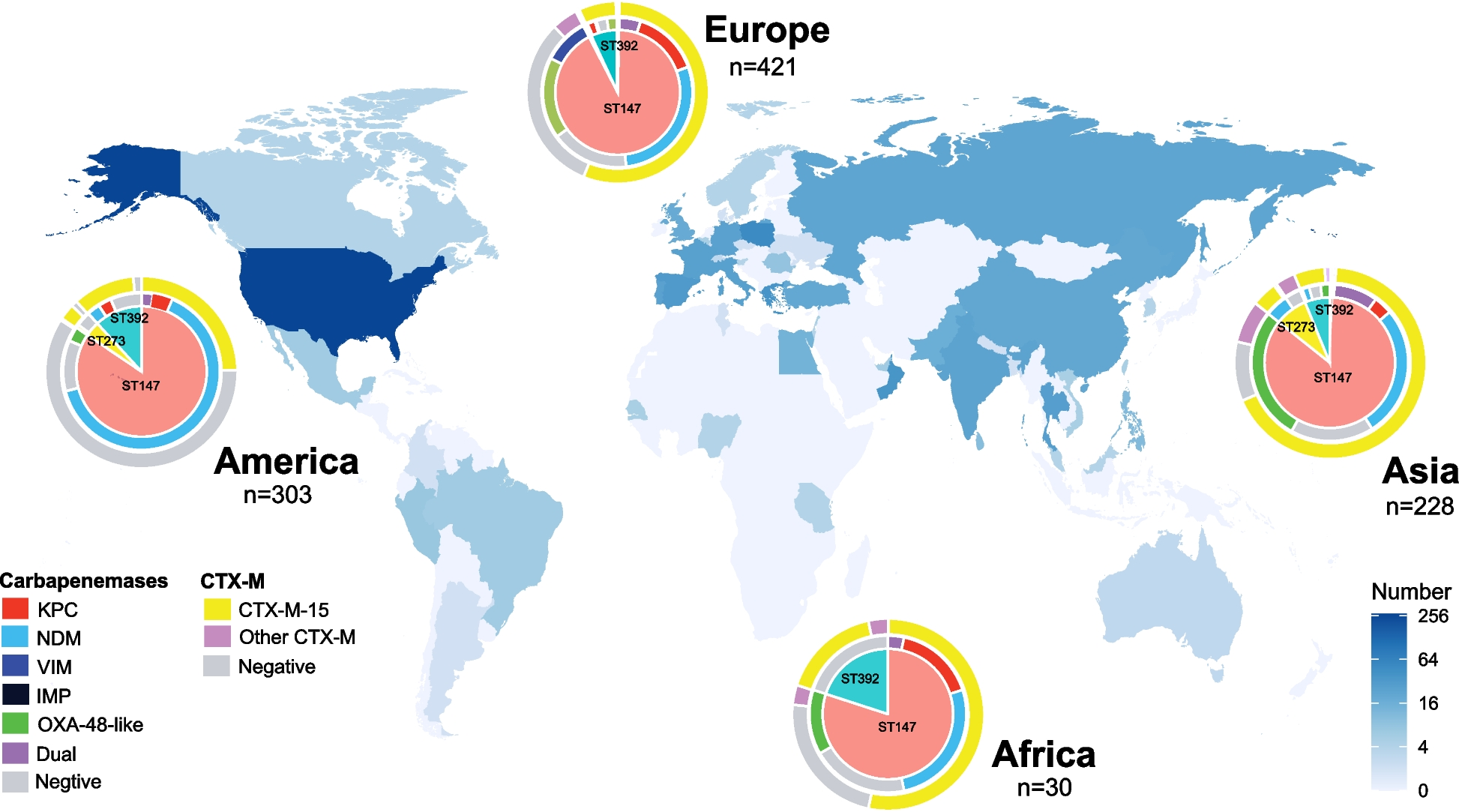








The 1012 strains originated from 59 different countries and spanned the period between 2002 and 2020. Isolates were collected from humans (n = 954, 94.3%), animals (n = 10, 1%), the environment (n = 14, 1.4%), and unknown (n = 34, 3.4%) sources. Isolates of human origin were obtained frequently from rectal swabs or stools (n = 302, 31.6%), followed by urine (n = 176, 18.5%), blood (n = 99, 10.4%), and the upper respiratory tract (n = 65, 6.8%). Most isolates were collected in North America (n = 278, 27.5%) and countries in Southern Europe (n = 241, 23.8%), Eastern Europe (n = 86, 8.5%), and Western Asia (n = 76, 7.5%) (Fig. 1). The major sequence types include ST147 (n = 881, 87.1%), ST392 (n = 85, 8.4%), and ST273 (n = 36, 3.6%) (Table 1).
Fig. 1
Isolate map depicting the collection sites of 1012 CG147 isolates over the world. Pie graphs show ST distribution in each continent, and donut graphs show carbapenemase (inner ring) and CTX-M (outer ring) distribution within each ST
Table 1 General characteristics of CG147 genomes publicly available from 2002 to 2020K. pneumoniae CG147 strains harbor AMR genes conferring resistance to multiple antibiotic families, including β-lactams, fluoroquinolones, aminoglycosides, rifampicin, tetracyclines, sulfonamides, and trimethoprim. Genes conferring resistance to tigecycline, colistin, and fosfomycin were less commonly observed (Additional file 1: Table S1). In this collection, we detected that 67.6% (n = 684) of isolates carried at least one CTX-M ESBL gene, with CTX-M-15 (n = 647, 94.6%) being the most frequent. Notably, 80.4% (n = 814) of isolates carried at least one carbapenemase gene. blaNDM was detected in 52.2% (n = 529) of isolates, blaOXA-48-like in 17.9% (n = 182) of isolates, blaKPC in 10.4% (n = 105), and blaVIM in 4.5% (n = 46) of isolates. Interestingly, in 2010, the frequency of KPC and NDM was similar; however, after 2014, NDM predominated among CG147 strains (Additional file 2: Fig. S1). The details of AMR genes, plasmids, capsular polysaccharide (CPS) types, and virulence factors are summarized in Additional file 3: Supplementary results.
The population structure and phylogenetic dating of CG147The CG147 phylogeny revealed a basal clade containing two quinolone susceptible isolates of ST147-KL55 and a larger clade containing the remaining isolates which carried QRDR mutations. In this larger clade, three primary branches emerged, corresponding to ST147 (n = 879, 86.9%), ST273 (n = 36, 3.6%), and ST392 (n = 85, 8.4%). Within the ST147 clade, two additional branches were identified. The first branch comprised 70 isolates, predominantly ST147 (n = 69, 98.6%) with one ST1880. Most of ST147 isolates within this branch encoded the capsular loci KL10 (66/69, 95.7%). These strains were collected in Southeastern Asia (27/69, 39.1%) and Western Asia (12/69, 17.4%), primarily from human hosts (66/69, 95.7%) and isolated from blood (17/69, 24.6%) and urine (15/69, 21.7%) specimens. The QRDR mutation GyrA-83Y-GyrA-87A-Par80I (67/69, 97.1%) was frequently found in this clade. Additionally, a small subclade within ST147-KL10 encoded yersiniabactin (ybt10-ICEKp4) (29/66, 43.3%) (Fig. 2).
Fig. 2
Phylogenetic analysis of 1012 genomes of CG147 strains. Colors in columns illustrate regions of origin, host, specimen, sequence type (ST), K locus, yersiniabactin, CTX-M, KPC, NDM, OXA-48-like, QRDR substitutions (GyrA and ParC), CRISPR-Cas type IV-A3 system, and anti-CRISPR proteins
The second branch consisted of ST147 isolates (n = 818) predominantly encoding the KL64 capsular locus (767/818, 93.8%). The most common QRDR mutation pattern among ST147-KL64 isolates was GyrA-83I-ParC-80I (711/767, 92.7%), followed by GyrA-83I-GyrA-87N-ParC-80I (48/767, 6.3%), with a small proportion carrying other mutations (8/767, 1%). Within this branch, four subclades were identified, correlated with different yersiniabactin lineages. The first subclade comprised 207 isolates encoding ybt9 in the ICEKp3 element (207/207, 100%). These isolates were exclusively recovered from human hosts (207/207, 100%), predominantly from Southern Europe (126/207, 60.9%), with rectal swabs or stool specimens being the most common sources (107/207, 51.7%). This subclade frequently carried β-lactamases such as CTX-M-15 (196/207, 94.7%) and NDM-1 (188/207, 90.8%), with no isolates encoding KPC carbapenemases (Fig. 2).
The second ST147-KL64 subclade carried ybt10 in the ICEKp4 element (69/72, 95.8%). These isolates were collected primarily in North America (14/72, 19.4%), Southern Europe (12/72, 16.7%), and Western Asia (12/72, 16.7%). Most were recovered from human hosts (61/72, 84.7%), with urine (16/61, 26.2%), and respiratory specimens (12/61, 19.7%) being the most common sample types. This subclade commonly encoded ESBL CTX-M-15 (69/72, 95.8%) and OXA-48-like carbapenemases (50/72, 69.4%), primarily OXA-181 (37/50, 51.4%). NDM variants, mainly NDM-5 (16/72, 22.2%) and NDM-1 (15/72, 20.8%), were also present, though only one isolate carried KPC-2 (Fig. 2).
The third ST147-KL64 subclade (n = 99) primarily encoded ybt16 in the ICEKp12 element (91/99, 91.9%). These strains were collected mostly in Eastern Europe (50/99, 50.1%), Southern Europe (8/99, 8.1%), and Western Europe (18/99, 18.2%) and were primarily of human origin (98/99, 98.9%). This subclade was frequently associated with CTX-M-15 (92/99, 92.3%) and NDM-1 (57/99, 57.6%), with some isolates also encoding OXA-48-like carbapenemases (32/99, 32.3%). None carried KPC carbapenemases, and CRISPR-IV-A3 was present in 17.2% (17/99) of isolates. A smaller subclade (n = 27) also carried ybt16 in ICEKp12 (26/27, 96.3%), with 12/27 isolates from human sources. This subclade encoded KPC-3 (24/27, 88.9%) but no other carbapenemases (NDM or OXA-48-like). Only three isolates (11.1%) encoded CTX-M-15 (Fig. 2).
The second main branch included all ST273 isolates (n = 36), frequently encoding KL74 (25/36, 69.4%). These strains were mostly collected in Southeastern Asia (10/36, 27.8%) and North America (9/36, 25.0%). CTX-M-type ESBLs were common (28/36, 77.8%), mainly CTX-M-15 (18/36, 50.0%). Yersiniabactin was rare among ST273 isolates, with 86.1% (31/36) testing negative. Only ten isolates carried NDM, two carried KPC-2, and another two had OXA-48 carbapenemases (Fig. 2).
The last main branch consisted of all ST392 isolates (n = 86), exclusively encoding KL27 (86/86, 100%). Most strains were from human sources (79/86, 91.9%), with specimen types including the urine (22/86, 25.6%), skin and soft tissue (13/86, 15.2%), respiratory (11/86, 12.8%), and blood (11/86, 12.8%). Yersiniabactin was rare in this branch, with 95.4% (82/86) testing negative; only two isolates carried ybt16-ICEKp12. CTX-M-15 was frequent (75/86, 87.2%), while carbapenemases were less common, with KPC detected in 17.3% (14/86), NDM-1 in 13.9% (12/86), and OXA-48-like in 13.9% (12/86) of isolates.
Three ST147-KL64 subclades from different regions independently acquired distinct yersiniabactin loci. In 2005 (95% CI, 2003–2007), a subclade primarily from Western Europe acquired ybt16; in 2009 (95% CI, 2009–2012), a Southern European subclade acquired ybt9; and in 2007 (95% CI, 2003–2009), a subclade from Western Asia and Eastern Europe acquired ybt10, suggesting geographically associated acquisition of yersiniabactin in CG147. The rmpADC and rmpA2 loci were estimated to have been acquired in 2002 (95% CI, 2000–2004) and 1998 (95% CI, 1994–2002), respectively.
We then implemented a Bayesian phylogeny analysis to reconstruct the global evolutionary history of CG147 using a subset of 306 genomes. The BactDating model estimates that the most recent common ancestor of CG147 was around 1963 (95% confidence interval [CI], 1948–1975) (Fig. 3). Our phylogenetic analysis identified an ancestral branch corresponding to the two ST147 isolates harboring KL55, which were predicted to be susceptible to most antibiotics, except for ampicillin, due to the chromosomal blaSHV-11 gene. Around 1990 (95% CI, 1985–1994), ST147 acquired two QRDR mutations (GyrA-83I and ParC-80I) and K locus (KL) 64, followed by acquisition of plasmids harboring resistance determinants, such as ESBLs and carbapenemases, and became an epidemic MDR cluster. Among this cluster, most strains were ST147-KL64; however, three major subclades evolved, including the emergence of ST273-KL74 in ~ 2003 (95% CI, 2000–2005), ST147-KL10 in ~ 2004 (95% CI, 2002–2007), and ST392-KL27 in ~ 2004 (95% CI, 2001–2006). Most of the CG147 strains (n = 884) harbored GyrA-83I and ParC-80I mutations; whereas, 48 strains harbored an additional GyrA-87N mutation. Interestingly, besides ParC-80I, the majority of ST147-KL10 strains (97.1%, 67/69) harbored the GyrA-83Y and GyrA-87A mutations, which appears to have originated by a genomic recombination event (see below). The detailed population structure of CG147 and phylogenetic dating of other genetic factors are described in Additional file 3: Supplementary results.
Fig. 3
Phylogenetic dating of 306 genomes of CG147 strains. Colors in columns illustrate regions of origin, host, specimen, sequence type (ST), K locus, yersiniabactin, CTX-M, KPC, NDM, OXA-48-like, QRDR substitutions (GyrA and ParC), type IV-A3 CRISPR-Cas system, AcrIE8.1, AcrIE9.2, and AcrIF11. Relevant evolutionary events are displayed in red boxes, and selected divergence time and 95% CIs are shown at the nodes
Capsular variation of CG147Our phylogenetic dating showed ST147-KL64 was likely descendant from ST147-KL55, while ST392-KL27, ST273-KL74, and ST147-KL10 originated from ST147-KL64 (Fig. 3). To unravel the recombination events associated with KL switching, we evaluated four major subclones with different ST, KL, and O locus combinations: i.e., ST147-KL64-O2v1, ST147-KL10-O3/O3a, ST392-KL27-O4, and ST273-KL74-O3b (Fig. 4A). The recombination analysis showed that all the KL-associated recombination was located in a ~ 793 Kb region (Fig. 4B) that spanned from a UbiX family flavin prenyltransferase gene (locus ID H1D29_06765 in Kp46564) to the MFS transporter gene (H1D29_12740) and encompassed the MLST locus of tonB, the fluoroquinolone-resistant-associated gene gyrA, and K and O antigen encoding operons. In detail, ST147-KL64-O2v1 resulted from a ~ 370 Kb recombination event that spanned from gltX (H1D29_08410) to hisA (H1D29_10050) between a ST147-KL55-O3a and a ST395-KL64-O2v1-like strain; ST392-KL27-O4 resulted from a ~ 558 Kb recombination event that spanned from bglX (H1D29_09590) and iron ABC transporter permease (H1D29_12350) between a ST147-KL64-O2v1 and a ST1758-KL27-O4-like strain; ST147-KL10-O3/O3a resulted from a ~ 305 Kb recombination event that spanned from UbiX family flavin prenyltransferase (H1D29_08925) and alpha/beta fold hydrolase (H1D29_10260) between a ST147-KL64-O2v1 and a ST3603-KL10-O3/O3a strain, and ST273-KL74-O3b resulted from a ~ 636 Kb recombination event that spanned from the ABC transporter substrate-binding protein (H1D29_09595) and MFS transporter (H1D29_12740) between ST147-KL64-O2v1 and a ST147-KL74-O3b strain (Fig. 4A). In addition, minor subclones including ST147-KL107-O2v1, ST147-KL81-OL101, and ST147-KL20-O2v1 were investigated, and they too resulted from recombination within the ~ 793 Kb region (Fig. 4B). An SNP density plot illustrated in Fig. 4C depicts the recombination regions.
Fig. 4
A Hypothesized evolutionary history in K. pneumoniae CG147 strains. B MLST allele locations on Kp46564 genome (GenBank CP059317). The blue arrow denotes the genome of Kp46564, and the green region shows the ~ 793-Kbp putative recombination region between the ST147 and other genomes. C Core genome SNP distributions of strains ST392-KL27, ST1758-KL27, ST147-KL10, ST3603-KL10, ST147-KL107, ST147-KL81, ST147-KL20, and ST147-KL55 compared to reference strain Kp46564. The number of SNPs (y axis) per 1000 nt is plotted according to the position on the Kp46564 genome (x axis). cps and lps are illustrated by small vertical bars in blue and green respectively. The box indicates the recombination region
CRISPR-Cas systems in CG147We next analyzed the distribution of CRISPR-Cas systems in 1012 CG147 strains. We identified two distinct CRISPR-Cas systems: a chromosome-borne type I-E system in 99.7% (n = 1009) and a plasmid-borne type IV-A3 system in 41.8% (n = 423) of the genomes.
A total of 96 different CRISPR spacers (short DNA fragments that are stored in CRISPR arrays for target recognition) were identified and 41 spacers were present in > 90% of the I-E CRISPR-Cas system harboring strains (Additional file 3: Supplementary results), indicating the I-E CRISPR-Cas system is conserved and an important element in the evolution of CG147.
To investigate the potential impact of I-E CRISPR-Cas system on the distribution of carbapenemase-encoding genes, we analyzed the presence of I-E protospacers (a complementary DNA sequence that is recognized by CRISPR-Cas system) in 1084 completely sequenced carbapenemase-encoding plasmids obtained from K. pneumoniae genomes in the NCBI database (accessed in 02/2023). Our investigation revealed the presence of 11 I-E protospacers on 423 carbapenemase-encoding plasmids. Notably, four protospacer sequences (IESP-2, [99.2%, 1001/1009], IESP-4, [99.1%, 1000/1009], IESP-30, [93%, 938/1009] and IESP-31, [92.6%, 934/1009], Additional file 1: Table S4) were identified highly prevalent in CG147 strains. The four highly prevalent protospacers were located within an approximate 830 bp region, including traC, a gene of DUF3560 domain, and the intergenic region between single-stranded DNA-binding protein (ssb) and SAM-methyltransferase gene on IncF plasmids. Notably, these protospacers-harbored IncF plasmids, such as IncFII(pHN7A8) (e.g., NZ_CP116905.1), IncFIB(pQil) (e.g., NZ_CP103551.1), IncFIB(Kpn3) (e.g., NZ_CP098346.1), IncFII(pKP91) (e.g., NZ_OW970445.1), or FII(pBK30683) (e.g., NZ_CP087613.1) plasmids, are frequently associated with KPC.
The IV-A3 CRISPR-Cas system was identified on IncHI1B/IncFIB plasmids in CG147 and most IV-A3 CRISPR-Cas system harboring isolates were collected from North America (n = 167, 39.5%) and Southern Europe (n = 146, 33.8%). These isolates clustered into two major subclades on the phylogenetic tree (Fig. 2). A total of 347 unique spacers were identified and only 11 spacers present in > 65% of the IV-A3 CRISPR-Cas harboring strains (Additional file 3: Supplementary results), suggesting that the acquisition of IV-A3 CRISPR-Cas system was a relatively recent event in comparison to the acquisition of I-E CRISPR-Cas system.
Among the 347 unique IV-A3 spacers, 16 were found to target 402 carbapenemase-encoding plasmids from the NCBI database. Among these, only one spacer (IVA3SP-10, [65.2%, 276/423], Additional file 1: Table S5) was present in > 65% strains, 13 spacers were harbored by > 2 strains, while the remaining were identified in individual strains. The mostly frequent encountered protospacer (IVA3SP-10) was located in traL on KPC- or CTX-M-harboring IncF plasmids, such as plasmids belonging to IncFII(pHN7A8) (e.g., NZ_CP107017.1), IncFIB(pQil) (e.g., NZ_CP027056.1), or FII(pBK30683) (e.g., NZ_CP059890.1) replicon types.
Among the 402 carbapenemase-encoding plasmids targeted by IV-A3 CRISPR-Cas system (from NCBI database), 332 were also targeted by I-E CRISPR-Cas system, indicating a redundant anti-IncF plasmid strategy by two different CRISPR-Cas systems in CG147.
Anti-CRISPR proteins in CG147We then mined the distribution of anti-CRISPR proteins in 1012 CG147 genomes. We identified anti-type I-E CRISPR protein AcrIE8.1 in 40.1% (n = 406), AcrIE9.2 in 54.2% (n = 548), and anti-type I-F CRISPR protein AcrIF11 in 3.2% (n = 32) CG147 strains (20.7% [n = 209] strains harbored both AcrIE8.1 and AcrIE9.2).
The gene acrIE8.1 was located on different prophages that size around 46 Kb. Two major prophage types were characterized: Phage-1 was in the arginine-tRNA ligase site on both the ST147 and ST273 chromosomes and Phage-2 was identified in ST392 where it inserts in the threonine-tRNA ligase site (Additional file 2: Fig. S3A). Phage-1 and Phage-2 are both intact prophages with defined attachment (att) sites, have high sequence similarity with each other, and likely evolved from a common ancestor (Additional file 2: Fig. S3A). The acrIE8.1 harboring strains were clustered into three major subclades on the phylogenetic tree, which corresponds to the presence of the two prophages (Additional file 2: Fig. S3B).
The genomic analysis showed that the acrIE9.2 was predominantly associated with a group of IncF plasmids, such as the epidemic blaKPC-harboring pKpQIL plasmids (e.g., CP059315, CP023928, and CP059315) in CG147 strains. Furthermore, the acrIF11 was identified on different IncF plasmids that lacked both I-E and IV-A CRISPR-Cas systems targeted sequences. These plasmids were more commonly harbored by E. coli (e.g. CP046417.1). The strains harboring acrIF11 are concentrated into one major cluster, all of which belonged to ST147 (Fig. 2).
The phylogenetic dating indicates that the chromosome-borne I-E CRISPR-Cas system emerged in CG147 as early as 1963 (95% CI, 1948–1975). The plasmid-borne IV-A3 CRISPR-Cas system was acquired around 1992 (95% CI, 1987–1996), which aligns with the emergence of anti-CRISPRs, including the phage-borne acrIE8.1 (1990 [95% CI, 1985–1994]) and the plasmid-borne acrIE9.2 (1992 [95% CI, 1987–1996]) in CG147. In contrast, the acquisition of acrIF11 appears to be a more recent event (2006 [95% CI, 2003–2009]).
Self-targeting CRISPR spacers in CG147We next analyzed the presence of self-targeting CRISPR spacers (additional copies of spacer sequences found elsewhere in the genomes but not in the CRISPR array) among CG147 strains. The existence of self-targeting spacers primarily indicates the functionality of anti-CRISPR mechanisms [54]. To avoid duplications, we included 646 non-redundant strains in this study, all of which possessed I-E CRISPR-Cas systems (Additional file 1: Table S6).
Interestingly, we found that a significant proportion (71.2%, 460/646) of strains contain I-E CRISPR-Cas self-targeting spacers. Among these, 218 harbored self-targeting spacers on plasmids, 87 on chromosomes, and 155 on both plasmids and chromosomes. Of the 242 strains harboring chromosomal self-targeting spacers, 87.6% (212/242) had self-targeting spacers located in prophage regions. This finding is unexpected because self-targeting spacers were expected to be eliminated by the endogenous I-E CRISPR-Cas system. However, further analysis revealed that nearly all the strains with I-E self-targeting spacers (442/460, 96.1%) also harbored at least one anti-type I-E CRISPR gene to overcome the presumed I-E immunity. This included 224 (48.7%) strains with acrIE8.1, 359 (78.0%) strains with acrIE9.2, and 141 (30.1%) strains possessing both. In contrast, the occurrence of anti-type I-E CRISPR proteins among strains lacking type I-E self-targeting spacers was considerably lower (2 strains with acrIE8.1 and 3 strains with acrIE9.2, 2.9%, 5/186) (p < 0.001). This significant difference strongly suggests that AcrIE8.1 and AcrIE9.2 provide protection against I-E CRISPR-Cas system, consistent with previous experimental tests [22, 23].
To further understand other factors that affect the presence of I-E self-targeting spacers, we conducted a logistic regression analysis of the presence of self-targets with STs, isolation regions, and the presence of anti-CRISPR proteins AcrIE8.1, AcrIE9.2, and AcrIF11. The analysis showed that only AcrIE8.1 (odd ratio [OR] = 537.98, p < 0.001) and AcrIE9.2 (OR = 977.55, p < 0.001) significantly associated with the presence of I-E self-targeting spacers (Table 2), providing additional credence to the hypothesis that AcrIE8.1 and AcrIE9.2 are countermeasures against the I-E CRISPR-Cas system.
Table 2 Logistic regression of type I-E and type IV-A3 self-targeting spacers with STs, regions, type IV-A3 CRISPR-Cas system, and anti-CRISPR proteins in 646 non-redundant strainsWe also examined the factors associated with the presence of IV-A3 CRISPR-Cas system. Among the 646 strains, 195 harbored the IV-A3 CRISPR-Cas system and ~ 60% isolates (116/195) possessed IV-A3 self-targeting spacers. Among them, 40 strains harbored self-targeting spacers on plasmids, 61 on chromosomes, and 15 on both plasmids and chromosomes, of which 46.1% (35/76) had self-targeting spacers located within prophage regions. However, the logistic regression analysis failed to detect any significant associations between the presence of anti-CRISPR proteins (AcrIE8.1, AcrIE9.2, and AcrIF11) and the occurrence of IV-A3 self-targeting spacers (Table 2), suggesting that these anti-CRISPR proteins may not have evolved as a countermeasure against the IV-A3 CRISPR-Cas system.
Associations between CRISPR-Cas systems, anti-CRISPR proteins, and AMR genes in CG147We next investigated the relationships between CRISPR-Cas systems, anti-CRISPR proteins, and AMR plasmids in CG147 at a population level (Fig. 5A). We employed a logistic regression analysis to examine the associations of AMR genes with STs, isolation regions, and the presence of IV-A3 CRISPR-Cas system, as well as anti-CRISPR proteins AcrIE8.1, AcrIE9.2, and AcrIF11.
Fig. 5
A Linking the interactions between CRISPR-Cas systems, anti-CRISPR protein, and AMR plasmids in CG147. B The frequencies of type I-E CRISPR-Cas targeted carbapenemase plasmids in CG147 during 2010–2018. C The frequencies of type IV-A3 CRISPR-Cas targeted carbapenemase plasmids in CG147 during 2009–2018
When both STs and geographic regions were controlled, we observed significant differences in the detection of the IV-A3 CRISPR-Cas system among various AMR strains. IV-A3 CRISPR-Cas system was commonly found in strains harboring NDM (OR = 4.16, p < 0.001), but less frequently associated with KPC-(OR = 0.52, p = 0.02) and CTX-M-positive strains (OR = 0.42, p < 0.001) (Table 3). To investigate this observation in more detail, we analyzed 50 completely assembled CG147 genomes including both chromosomes and plasmids. The result showed that the primary target of the IV-A3 CRISPR-Cas system, traL, was consistently present in plasmids associated with KPC and CTX-M production (e.g., CP023916, CP075261.1, and CP023928), but absent in plasmids linked to NDM or OXA-48-like genes. Furthermore, we noted instances where blaNDM-1 co-existed with the IV-A3 CRISPR-Cas system on the same plasmids (e.g., CP066856). This co-occurrence may contribute to the increased association between NDM and the IV-A3 CRISPR-Cas system.
Table 3 Logistic regression of KPC, NDM, OXA-48-like and CTX-M with STs, regions, type IV-A3 CRISPR-Cas system, and anti-CRISPR proteins in 646 non-redundant strainsThe result analyzing the prevalence of the anti-CRISPR proteins showed a positive association between AcrIE8.1 and NDM (OR = 4.16, p < 0.001) and CTX-M (OR = 2.33, p < 0.001), but a negative correlation with KPC (OR = 0.48, p = 0.02). A more in-depth analysis of the association between KPC subtypes and AcrIE8.1 (Additional file 1: Table S7) revealed that the negative correlation was primarily driven by the over-representation of KPC-3 in AcrIE8.1-negative CG147 strains (OR = 0.04, p = 0.004). Notably, common blaKPC-3-harboring IncN plasmids (similar to LT838197) were detected in most KPC-3-producing isolates from Portugal and France. Of significance, these plasmids do not possess the I-E protospacers.
Additionally, our results unveiled a significantly positive association between AcrIE9.2 and KPC (OR = 3.18, p < 0.001), particularly with KPC-2 (OR = 4.77, p < 0.001), while no such association was observed with NDM, OXA-48-like, or CTX-M. Through the analysis of complete genome sequences, we have identified the co-existence of acrIE9.2 with blaKPC-2 on certain plasmids, notably the epidemic pKpQIL plasmids (e.g., CP059315, CP023928, and CP059315). Interestingly, as aforementioned, pKpQIL-like plasmids also harbor I-E and IV-A3 CRISPR-Cas systems target sequences, illustrating a unique example of the co-occurrence of AMR genes, the CRISPR-Cas targets, and anti-CRISPR genes in the same epidemic plasmid vector.
Lastly, we evaluated the proportions of CRISPR-Cas system-targeted carbapenemase plasmids over time. Our findings indicate that the frequencies of I-E CRISPR-Cas (Fig. 5B) and IV-A3 CRISPR-Cas systems that targeted (Fig. 5C) carbapenemase plasmids declined year by year from 2009 to 2019.
Experimental verification of the function of CRISPR-Cas systems and anti-CRISPR proteins from CG147To assess the anti-plasmid capability of the I-E CRISPR-Cas system, we conducted a conjugation assay comparing the transfer frequency of the pKpQIL plasmids, both with and without type I-E CRISPR-Cas targets, into strain BK56682. BK56682 is an ST147 strain, possessing the I-E CRISPR-Cas system but it lacks the IV-A3 CRISPR-Cas system and acr genes. For this investigation, the KPC-encoding pKpQIL plasmid was selected as the test plasmid as it is an epidemic blaKPC-harboring vector in various Enterobacteriale species, and it contains targets for both I-E and IV-A3 CRISPR-Cas systems. Specifically, we tested plasmid pKpQIL-03, which carries the four highly frequent I-E protospacers. It is important to note that the native pKpQIL-03 also contains acrIE9.2, and therefore, to avoid any potential confounding effects from this anti-CRISPR, we knocked out acrIE9.2 on pKpQIL-03 (designated as pKpQIL-03ΔacrIE9.2) in the test plasmid. Subsequently, we knocked out the 1315 bp region containing the four common type I-E protospacers on pKpQIL-03ΔacrIE9.2 (designated as pKpQIL-03ΔacrIE9.2ΔIEtarget). The results demonstrated that the conjugation frequency of pKpQIL-03ΔacrIE9.2 reduced by approximately 7000 times (p < 0.001) compared to pKpQIL-03ΔacrIE9.2ΔIEtarget (Fig. 6A), confirming the anti-plasmid functionality of the I-E CRISPR-Cas system in CG147.
Fig. 6
A Conjugation frequencies of pKpQIL-03△acrIE9.2 and pKpQIL-03△acrIE9.2△IEtarget to BK56682. B Conjugation frequencies of pKpQIL-03△acrIE9.2 transferred to BK56682::pClone and BK56682::pClone-acrIE8.1. C Plasmid stabilities of pKpQIL-03△acrIE9.2 in BK56682::pClone and BK56682::pClone-acrIE8.1. D Conjugation frequencies of pKpQIL-03△acrIE9.2 transferred to BK56682::pClone and BK56682::pClone-acrIE9.2. E Conjugation frequencies of pKpQIL-03△acrIE9.2 transferred to DH5a::pClone, DH5a::pClone-IVA3-1 (no transconjugant observed) and DH5a::pClone-IVA3-2 (no transconjugant observed). TC transconjugants, R recipient strains. *p < 0.05, **p < 0.01, ***p < 0.001
We then evaluated the anti-CRISPR efficacy of AcrIE8.1 against the I-E CRISPR-Cas system in CG147. The gene acrIE8.1 was cloned into plasmid vector, pClone, and transferred to BK56682. This was followed by a conjugation assay comparing the transfer frequency of the pKpQIL plasmid into BK56682, both with (pClone-acrIE8.1) and without (pClone) acrIE8.1. The conjugation assays revealed that the transfer frequency of pKpQIL-03ΔacrIE9.2 to BK56682 with pClone-acrIE8.1 was 4.2 times higher (p < 0.05) than to BK56682 with the control plasmid (Fig. 6B), thus confirming the anti-CRISPR effect of AcrIE8.1.
To assess the dynamics of the I-E CRISPR-Cas system over time, we conducted plasmid stability experiments and compared the results of serial passaging the strains. The findings showed that the carriage rate of pKpQIL-03ΔacrIE9.2 remained around 80% after 60 passages in strains with a repressed I-E CRISPR-Cas system, while the carriage rate dropped to approximately 10% in negative control strains after 20 passages (Fig. 6C), confirming the plasmid immunity of the I-E CRISPR-Cas system.
We also explored the anti-CRISPR effect of AcrIE9.2 by conjugating pKpQIL-03ΔacrIE9.2 to BK56682 with (pClone-acrIE9.2) and without acrIE9.2 (pClone control). The conjugation frequency of pKpQIL-03ΔacrIE9.2 in BK56682::pClone-acrIE9.2 was 3.7 times higher (p < 0.001) compared to the transfer in BK56682::pClone, indicating that AcrIE9.2 also possesses an anti-CRISPR effect (Fig. 6D).
To verify the functionality of the plasmid-borne IV-A3 CRISPR-Cas system, we cloned two different IV-A3 CRISPR-Cas arrays (both with spacers targeting pKpQIL-03) into pClone (pClone-IVA3-1 and pClone-IVA3-2) and transferred them to E. coli DH5a. Through conjugation assays, we observed that the conjugation frequencies of pKpQIL-03 transferred to DH5a::pClone-IVA3-1 and DH5a::pClone-IVA3-2 were approximately 900 times lower (p < 0.001) than that of pKpQIL-03 transferred to DH5a with the control plasmid (Fig. 6E), indicating the robust anti-plasmid function of the IV-A3 CRISPR-Cas system. Lastly, despite the detection of AcrIF11 in our experiments, no significant anti-CRISPR effects on the I-E and IV-A3 CRISPR-Cas systems were observed (data not shown).
留言 (0)