
記住我


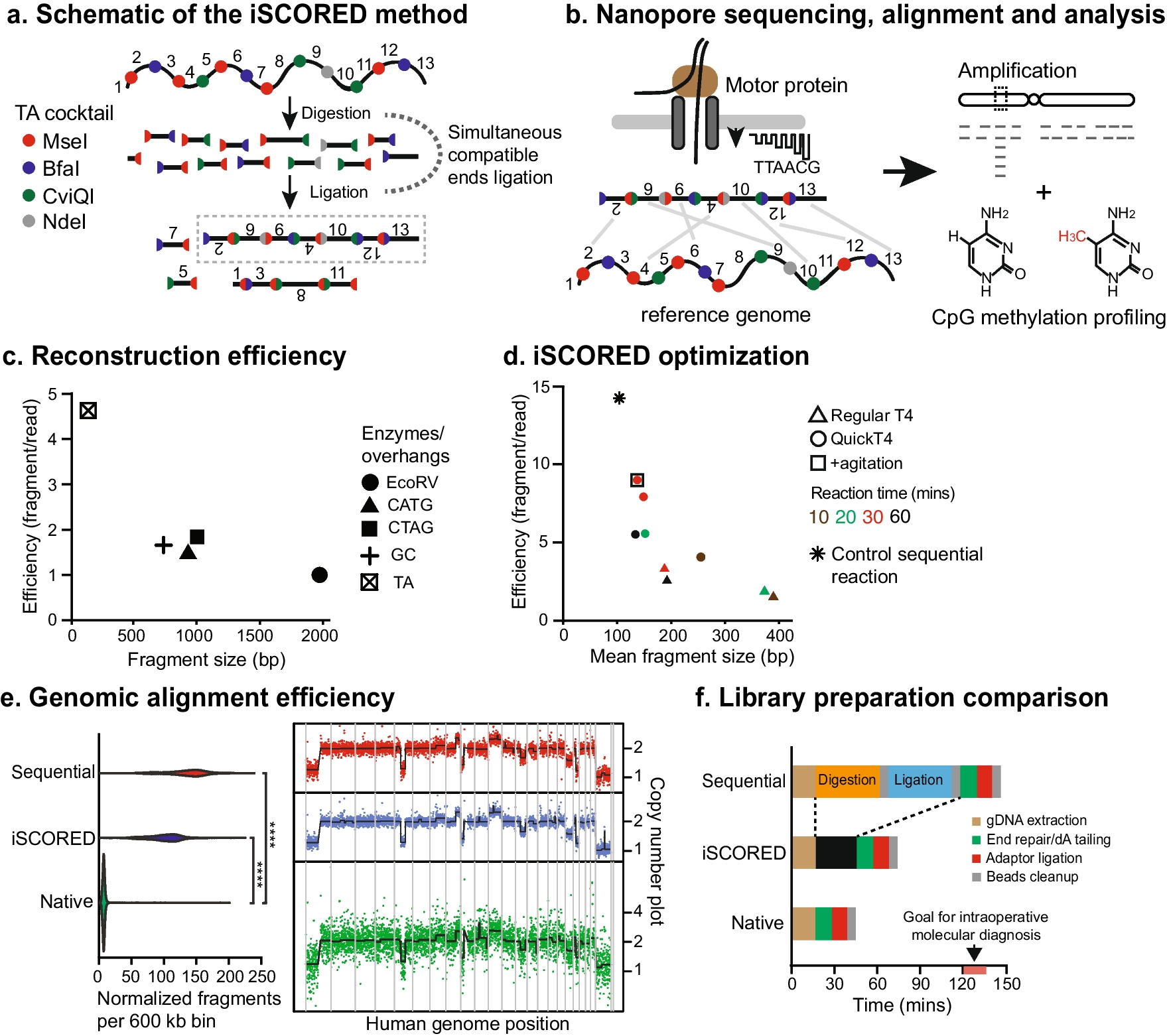






The central concept of iSCORED is simultaneous digestion and ligation of DNA molecules by utilizing a panel of restriction endonucleases (REs) capable of generating compatible cohesive ends. Within the same reaction, DNA ligase catalyzes random re-ligation of the digested fragments to form long concatemers. Irreversible ligation products are generated when cohesive ends produced by different restriction enzymes are ligated together. This unidirectional reaction is possible because of the staggered nature of the DNA recognition sequences and actual phosphodiester bond breakage sites (Fig. 1a, b, Additional file 1: Fig. S1). The likelihood of forming such irreversible ligations increases with the number of different restriction enzymes producing compatible ends. Using CTAG overhangs as an example, the digested fragments were concatenated to larger chimeric molecules in the presence of DNA ligase (Additional file 1: Fig. S1).
Fig. 1
Proposed iSCORED method for rapid copy number analysis. a iSCORED schematic showing simultaneous compatible end ligation with TA enzyme cocktail (T^TAA, C^TAG, G^TAC, and CA^TATG by MseI, BfaI, CviQI, and NdeI, respectively). b Long stochastically concatenated DNA molecules are analyzed with a Nanopore device and aligned to the reference for genome-wide quantitative measurement. c The reconstruction efficiency of four iSCORED cocktail combinations is compared. The reaction is incubated at 37 °C for 30 min. CATG cocktail: NcoI (C^CATGG), PciI (A^CATGT), BspHI (T^CATGA). CTAG cocktail: NheI (G^CTAGC), SpeI (A^CTAGT), AvrII (C^CTAGG), XbaI (T^CTAGA). CG cocktail: MspI (C^CGG), HinP1I (G^CGC), HpyCH4IV (A^CGT), and TaqI-V2 (T^CGA). EcoRV is employed as a control since it generates blunt ends upon restriction digestion. d Optimization of iSCORED reaction by adjusting various experimental parameters, such as incubation periods, DNA ligases, and intermittent mixing and cooling. e An oligodendroglioma sample was processed either sequentially (digestion, purification, and ligation), with iSCORED, or sequenced as native gDNA. Samples were normalized to contain the same amount of sequencing data. The number of unique fragments mapped per genomic bin are shown for each sample (left panels). The resulting CNV plots are shown in the right panels (resolution = 600 kb per bin). CoV for sequential approach, iSCORED, and native gDNA sequencing are 0.57, 0.54, and 3.3, respectively. f Comparison of library preparation times across three methods. The sequential DNA digestion and ligation required 150 min, while the iSCORED required 75 min and the native DNA method required 45 min. The goal of intraoperative molecular diagnosis is achieved within 120–150 min of receiving the resected specimen
Systematic analysis and optimization of all overhang candidatesWe next examined all existing 4-mer and 6-mer Type IIP REs capable of generating 2-nucleotide and 4-nucleotide overhangs (Additional file 1: Fig. S2). Given the palindromic nature of Type IIP REs, there are 16 (= 42) and 4 (= 41) possible combinations for 4-nucleotide and 2-nucleotide overhangs, respectively. Depending on the RE recognition sequence (4 or 6 bp), the same overhang could be generated by 41 or 42 different enzymes; however, some of the theoretical combinations do not exist, and some are partially or completely blocked by DNA methylation (Additional file 1: Fig. S2; New England Biolabs).
We tested the top four overhang candidates that had the highest number of RE combinations while exhibiting the least possibility of methylation inhibition (Fig. 1c). To quantitatively measure the reconstruction efficiency (i.e., the number of uniquely mapped fragments per sequencing read), we compared combinations generating 4-nt overhangs with those generating 2-nt overhangs. Surprisingly, we found that the efficiency of 4-nt overhang combinations is not superior to that of 2-nt overhang combinations (Fig. 1c, Additional file 1: Fig. S3). This is presumably due to the significantly higher number of generated fragments by REs with 2-nt overhangs. The most efficient combination was the TA overhang cocktail mix that consisted of MseI, BfaI, CviQI, and NdeI, resulting in a mean reconstruction efficiency of 4.6 and mapped fragment of 120 bp. To further optimize the iSCORED reaction, we tested various incubation periods and DNA ligases (Fig. 1d). Our experiments revealed that an incubation period of 30 min at 37 °C with intermittent agitation at 18 °C (900 rpm) yielded the highest mean reconstruction efficiency of 8.7 (Fig. 1d). This experimental condition was thus utilized for the remainder of the study.
Genomewide aneuploidy detection in tumorsTo detect large-scale CNV (> 10 Mb) and aneuploidy, the sequenced reads were first segmented into individual fragments by identifying short matches when mapping to the GRCh37/hg19 reference genome [38]. These uniquely mapped fragments were then filtered for quality (alignment scores ≥ 120, see the “Methods” section for details) and assigned to predefined genomic bins (600 kb) for quantitative analysis. High numbers of mapped fragments per bin generated low variability between bins and this helped ensure high confidence in the resulting CNV plot. Finally, circular binary segmentation [49] through DNACopy [50] was employed to identify copy number alterations across genomic bins.
The performance of the iSCORED pipeline was compared to the conventional sequential SMURF approach [38] (i.e., digestion, purification, and ligation) and unprocessed native gDNA. By normalizing the datasets to the same amount of total DNA sequence, both the iSCORED and SMURF methods exhibited an over 16-fold increase in the number of fragments compared to native gDNA sequencing (Fig. 1e). The significant increase in fragment count resulted in low variability, which was critical for detecting copy number changes with high confidence. Specifically, the coefficient of variations (CoV) for the SMURF, iSCORED, and native gDNA sequencing were 0.54, 0.57, and 3.3, respectively. Detection of large CNV and aneuploidy by iSCORED showed 100% concordance with clinically validated chromosomal microarray data (Additional file 2: Fig. S9) and also demonstrated a much higher resolution than short-read based analysis [36] within a comparable timeframe of 2 h (Fig. 1f, Additional file 1: Table S2) [51]. While the SMURF approach demonstrates a slightly better reconstruction efficiency (14.5), its sequential approach requires a substantial input DNA (2–3 μg) [38] and an extended preparation time (90–120 min) [38, 39]. This is in contrast to the iSCORED method, which requires only 200–400 ng of input gDNA and a preparation time of 30 min. The prolonged process prohibits its intraoperative application in standard CNS surgeries (typically 3–4 h; Additional file 1: Table S3). The molecular information is crucial for accurate pathology diagnosis as many primary CNS tumors are defined by molecular alterations. For instance, chromosomal 7 gains and 10 losses are characteristic of glioblastoma, while the presence of 1p/19q codeletion is required for oligodendroglioma diagnosis according to the 2021 WHO Classification [13].
Refinement of quantitative measures to detect copy number variationsWhile large 600-kb bins are effective in detecting aneuploidy, most clinically-relevant gene amplifications occur within a range of hundreds of kilobases to a few megabases [52, 53]. In such cases, using a large bin could result in averaging out the dose change, leading to decreased accuracy of small amplifications. Thus, we refined the bin size to 60 kb, which was similar to the highest genomic resolution of clinically validated chromosomal microarray analysis (CMA). When using the control human genome (NA12878) to quantify the total mapped fragments in refined 60-kb bins, the numbers mapped fragments fluctuated substantially across the predefined bins (Fig. 2a). Since the iSCORED is a restriction enzyme-based method, this finding was presumably due to variations in the density and distribution of restriction enzymes’ cutting sites across the human genome. We have termed the phenomenon intrinsic regional variability (IRV) (Fig. 2a, Additional file 1: Fig. S4). The background fluctuations might allow for tolerating outliers driven by true copy number changes, impacting the detection accuracy. In addition, this fluctuation behavior was inversely related to the amount of the data acquired (Fig. 2b). Hence, this finding was characterized in the context of the corresponding number of fragments in the genome.
Fig. 2
Normalization of variable mapped fragments in predefined bins for accurate copy number detection. a The number of mapped fragments per bin fluctuates across the wild-type genome (intrinsic regional variability, IRV), yielding a relatively high coefficient of variation (CoV) of 0.68 and hampering detection of true outliers. b Extensive sequencing does not address the fluctuation due to IRV (left panel). Normalizing the samples with the control wild-type dataset, the CoV dramatically drops and stabilizes at ~ 1 million mapped fragments (right panel). c The control genome data displayed CoV of 0.09 after normalization (upper panel). Application of this approach allows for detecting regions of amplification in both chromosome 2 and chromosome 19 (defined as copy number > 10). d Mixture of tumor with wild-type gDNAs shows that the amplified copies increase as the tumor percentage increases (Pearson correlation coefficient of 0.99). Using Z values of 10 as cutoff, the genetic amplification CCNE1 in chromosome 19 could be reliably detected at 5% tumor purity with 500,000 mapped fragments
For this purpose, we utilized the coefficient of variation (CoV) [54] as a quantitative index and performed a time-lapsed analysis of the sequenced control samples. After comparing the sub-datasets with varying numbers of mapped fragments, we found that significant fluctuations reached a plateau around a CoV of 0.68 at one million mapped fragments, regardless of extensive sequencing (Fig. 2b, left panel). To address this issue, we performed bin-specific normalization by calculating a ratio of the mapped fragments in the sample of interest to those in the commonly used control reference genome (NA12878). This normalization significantly reduced the observed genomic fluctuations by approximately fourfold (Fig. 2b, right panel). The CoV of the normalized data was substantially reduced to 0.09 down from 0.68 in the corresponding non-normalized data.
This normalization process also allowed us to infer the required number of total mapped fragments to reliably identify regions of copy number change. When investigating the slope [55] of CoV as a function of the acquired fragments, the inflection point was at a datapoint with mapped fragments of < 500 k, while the first order derivative function approached a value of zero at about one million mapped fragments (Fig. 2b, right panel). Thus, we determined that acquiring approximately one million mapped fragments was sufficient to reliably detect genomic dosage changes. It is worth noting that pre-defined bins with inherently low counts can lead to high sampling variability, resulting in false positive detections. Using normal well-characterized control gDNAs (NA20967, NA12878, NA24385, and NA24631 from Coriell Institute), we established that excluding bins within the lowest 0.2% genomic counts ensures reliable genomic dosage assessment (Additional file 1: Fig. S4). Bin-specific normalization and lowest 0.2% bin exclusion helped effectively detect true copy number variations by minimizing the effects of intrinsic regional variability. Finally, with the advent of a complete human genome reference that includes pericentromeric and subtelomeric regions [56, 57], we have aligned our iSCORED datasets to the T2T reference genome (Additional file 2: Fig. S9, see the “Methods” section for details) [42].
Gene amplification across various tumor purity levelsTo demonstrate the effectiveness of the iSCORED procedure and its analysis pipeline, we first analyzed a metastatic adenosquamous carcinoma from the esophagus (Case M9 in Table 1). A CCNE1 gene amplification was detected in chromosome 19 (140 copies, inset in Fig. 2c), consistent with clinically validated next-generation sequencing. We further determined the minimum tumor percentage to reliably detect CCNE1 amplification by assessing a range of mixtures comprising control gDNA (NA12878) and tumor gDNA. This revealed a positive correlation between increasing amplification and rising tumor percentage (Pearson r = 0.99). By utilizing a Z-score cutoff of 10 to establish detection confidence, we were able to detect CCNE1 amplification in samples with as low as 5% tumor percentage using only 500 k mapped fragments (Fig. 2d). Additionally, low copy number gain (22 copies) was also reliably detected with the same parameter, albeit at a higher tumor percentage and with more fragments (20% and 1.5 million fragments, respectively, Additional file 1: Fig. S5). Overall, our results demonstrate the effectiveness of the iSCORED pipeline in detecting gene amplifications, even in samples with low tumor purity. The detection resolution outperforms the tumor percentage thresholds employed by clinical next-generation sequencing platforms, which are generally set at 15–20%.
Table 1 Comparison of oncogene amplification results between iSCORED and NGSRapid molecular analysis of the brain tumor cohortTo validate the iSCORED pipeline, we performed blind testing of a cohort of 26 intracranial neoplasms, including 17 primary CNS tumors and 9 metastatic tumors. Taking advantage of the mechanical destruction of frozen tissue sectioned at 5 μm thickness using a cryostat, high-quality gDNA is extracted within 15 min and processed through the iSCORED pipeline (see methods for details). The performance was timed and the findings were compared to the results from clinically validated next-generation sequencing (TruSight® Tumor 170 and whole exome sequencing) and chromosomal microarray analysis (Affymetrix OncoScan®) [40].
Within 1-h of MinION sequencing, an average 344 ± 24 Mb of data were generated, corresponding to 1.38 ± 0.08 million mapped fragments (SEM, Additional file 1: Fig. S6) [51]. This is higher than the predetermined required data quantity for confident CNV detection (0.5–1 × 106 mapped fragments, Fig. 2b). Furthermore, the output is approximately threefold the data volume of the recently published SMURF-based method (nCNV-seq) within the same 50–60 min sequencing window [39]. Across the 26 investigated samples, the diagnostic accuracy of the iSCORED platform was 100% in detecting gene amplification of more than 10 copies (95% confidence interval [58]: 91–100%, Pearson r = 0.81 by comparing to the NGS results; Table 1). One sample was detected to have MYB amplification (21 copies; case M8) by the iSCORED pipeline, a finding that was not originally uncovered by TST 170 panel but was later verified by a whole exome NGS study (13 copies).
The output genomic graph from the iSCORED pipeline provided precise information on amplified regions and the confidence of detected outliers (Fig. 2c and Additional file 2: Fig. S9). EGFR amplification is a molecular defining alteration in glioblastomas, typically occurring as extrachromosomal DNA ranging from 1–3 megabases (Mb) in size [52, 59]. In our cohort of six EGFR-amplified glioblastomas, the average amplification regions spanned 1.66 ± 0.44 Mb (SEM) with an average copy number of 150.5 ± 47 (SEM). These samples also exhibited diverse regions and degrees of amplification, which is consistent with the known heterogeneity of glioblastoma [60, 61] (Fig. 3d).
Fig. 3
Concurrent methylation analysis of primary CNV tumors. a Acquired methylation classification features with iSCORED-processed MinION sequencing over time using the Sturgeon and Rapid CNS. b Comprehensive comparison of Rapid CNS and Sturgeon methylation classification for primary brain tumors across multiple time points from the initiation of sequencing. c In silico mixture of glioblastoma, medulloblastoma, and oligodendrogliomas with control brain tissue dataset at various ratios using the Sturgeon (T2T) and Rapid CNS (hg38) (total data quantity after 1 h of sequencing). d Exact amplified regions covering EGFR oncogene in glioblastoma samples. e Methylation characterization of amplified EGFR oncogene reveals promoter hypomethylation. Subep = subependymoma, oligo = oligodendroglioma, medullo = medulloblastoma, GBM = glioblastoma
Concurrent tumor methylation classification using iSCORED-generated datasetMethylation classification of tumor types has emerged as an important diagnostic tool in clinical practice, particularly in brain tumors [9, 12]. The Heidelberg methylation classifier, for instance, successfully classified 91 tumors from 2801 formalin-fixed paraffin-embedded (FFPE) archival tissue [9]. Leveraging the capability of Nanopore sequencing to identify 5-methycytosine (5mC) from native DNA without additional sample preparation, we extracted methylation information from our sequencing data and classified it using Rapid-CNS2 and Sturgeon, both of which are machine learning classification systems trained on the Heidelberg dataset [10].
To evaluate the reliability of methylation classification over time, we first processed MinION data at five timepoints (10, 20, 30, 45, and 60 min). RapidCNS2 extracted the number of methylation features that overlapped with the 100 k most variable features from the Heidelberg dataset (Fig. 3a). Within 45 min, all samples had identified more than 1,000 CpG features, reaching the cut-off determined by the RapidCNS2 authors [62], and 10 out of 14 classification results aligned with the final pathological diagnoses (Fig. 3a, b and Additional file 1: Fig. S7). While Sturgeon utilizes all 450 k features from the Heidelberg dataset, leading to a larger overall number of features, these samples also reached sufficient CpG sampling within 45 min, and achieved concordant diagnoses in 10 out of 14 cases. In comparing the concordant cases, Sturgeon (T2T and hg38 reference) achieved a passing score (0.8) in 9 out of 10 cases, whereas Rapid-CNS2 (hg38 reference) achieved a passing score (0.6) in 8 out of 10 cases. Among the 4 inconclusive cases, Sturgeon classified two cases (GBM3 and GBM4) as control inflammation with high calibrated scores (0.97–1.0), whereas Rapid-CNS2 assigned lower scores to the same cases (0.28, 0.27 for GBM3 and GBM4, respectively) (Fig. 3a and b, Additional file 3: Table S6).
Rapid-CNS2 and Sturgeon utilized datasets derived from native DNA Nanopore sequencing, which are long and unfragmented. To validate that our short concatenated fragments generated via iSCORED did not affect methylation classification, we compared the data of three oligodendroglioma samples processed with iSCORED to those from native DNA sequencing using both Rapid CNS2 and Sturgeon systems [12, 45]. The results revealed comparable classification scores, suggesting that fragmentation in iSCORED did not appear to affect the accuracy of methylation classification. Of note, while the oligodendroglioma with a lower tumor percentage (80.5% in oligo_1, Additional file 1: Fig. S7a) consistently fell below the 0.6 threshold in Rapid CNS2, all three samples passed the required threshold (0.8) in the Sturgeon (T2T) system after 30 min of MinION sequencing (Additional file 1: Fig. S7c).
Tumor purity can significantly affect methylation classification results [9, 12]. This effect is particularly pronounced in glioma due to their infiltrative growth pattern, resulting in a mixture of neoplastic cells, normal brain parenchyma, and inflammatory cells. By comparing to final pathological diagnoses, our data is consistent with this observation: all four samples showing inconsistent classification at 60 min of sequencing were gliomas with low tumor percentages (20–60%). To further assess the impact of tumor purity on classification accuracy, we in silico admixed data from three tumors with the highest classification scores with control CNS tissue (frontal cortex, Fig. 3d). Our analysis revealed a drop in classification scores as tumor purity decreased, with Sturgeon analysis being more resistant to low tumor purity (Fig. 3c). However, both RapidCNS2 and Sturgeon either fell below the classification threshold (0.6 and 0.8, respectively) or did not yield consistent classification results when the tumor purity was 60% or lower. Overall, Sturgeon analysis (T2T reference) exhibits a superior calibrated classification score over the course of sequencing (Fig. 3b) and higher resistance to tumor purity issue when compared to Rapid-CNS2 (hg38). We have thus selected Sturgeon (T2T) as the methylation classification neural network for intraoperative molecular diagnosis.
Promoter hypomethylation in the amplified oncogenesThe epigenetic landscapes of amplified oncogenes offer mechanistic insights into transcriptional regulations [52, 63]. Despite the inherent low-pass nature of iSCORED, gene amplification ensures sufficient coverage for methylation profiling in the defined regions. Using glioblastoma as a proof of principle, within 1 h of MinION sequencing, we consistently detected hypomethylation across approximately 285 CpG sites within the promoters of the amplified EGFR (n = 6, coverage depth of 7.6 ± 2.7 (SEM), Fig. 3e, f). Such promoter hypomethylation is not exclusive to amplified EGFR of glioblastoma. Low 5mC percentages at CpG islands of oncogene promoters were detected in two other amplified oncogenes in glioblastomas (MYCN in GBM1 and MDM2 in GBM6), as well as five oncogenes amplified in metastatic tumors (FGFR1 and CCND1 of breast cancer in M1, ERBB2 of lung cancer in M6, MYB of esophageal cancer in M8 and CCNE1 of esophageal cancer in M9; Additional file 2: Fig. S9).
Intraoperative validation of 15 diagnostically challenging CNS tumorsWe employed the iSCORED pipeline for rapid molecular characterization of a prospective cohort of diagnostically challenging primary brain tumors during craniectomy at DHMC. The study included 8 high-grade gliomas, 4 low-grade gliomas, and 3 spindle cell neoplasms (Fig. 4b). The average time from specimen arrival to completion of DNA extraction was 20 ± 0.2 min (SEM). The iSCORED procedure consistently took 30 min, and subsequent library preparation before loading to PromethION flowcells took 31.8 ± 0.5 min (SEM). Since the first 5–6 min of Nanopore PromethION sequencing is used for flowcell check rather than library sequencing [12], we coordinated with library preparation to load the DNA library at the end of the flowcell check to avoid delays in data generation. The bioinformatics pipeline was programmed to analyze the initial 18 min of generated data [42], aiming to achieve 0.5–1.0 × 106 mapped fragments. Final data analysis and integration, including methylation classification and copy number plot generation, took 4.9 ± 0.4 min (SEM). The entire workflow, specimen arrival to output graph generation, was accomplished within 104.7 ± 0.7 min (SEM). The PromethION flowcells were used up to 7 times and generated 168 ± 15 Mb of data within 18 min, which corresponded to an average of 708,163 ± 77,700 (SEM) mapped fragments and identified 19,296 ± 1571 methylation features (SEM) [42, 51] (Fig. 4c).
Fig. 4
Prospective molecular analysis of diagnostically challenging brain tumors with iSCORED pipeline. a Shown is the incorporated iSCORED workflow applied during intraoperative morphology-based diagnosis. Additional 10–15 scrolls of tissue sections, each 5 μm thick, are prepared to extract gDNA for subsequent iSCORED library preparation. Either MinION or PromethION sequencing could be utilized (both with concurrent analysis during sequencing). The final output graphs comprise whole genome CNV, gene amplification regions, and methylation classification with quantitative confidence scores (Z scores for gene amplification and calibrated scores for methylation classification). b Real-time intraoperative molecular diagnosis with precise timestamps recorded from tissue arrival to final reports in 15 diagnostically challenging brain tumors. The entire workflow could be completed within ~ 105 min. The morphology-based intraoperative diagnosis was compared to generated molecular results, including methylation classification and CNV results [51]. The numbers within the brackets of methylation classification and oncogene amplification denote the calibrated scores of corresponding diagnoses and detected copy number, respectively. * Scores of different glioblastoma subtypes. PXA = pleomorphic xanthoastrocytoma c Sequencing data, including mapped fragments and identified CpG sites, obtained within the initial 18 min (PromethION flowcells). d A comparative analysis of Nanopore-based molecular assays, including genomic detection resolution, library preparation time, input genomic DNA quantity, and required sequencing duration, reveals the iSCORED-based assay as the only method to achieve genome-wide high-resolution CNV detection within the surgical window. SMURF = sampling molecules using re-ligated fragments [38]. STORK = short-read transpore rapid karyotyping [36]. WGS = whole genome sequencing [35]
In the cohort, the cases were selected due to the pressing need to define glioblastoma within the high-grade glioma category. Among the high-grade gliomas, methylation profiling based on the Sturgeon (T2T reference) method accurately classified 7/8 as glioblastoma, with calibrated scores of 0.94 ± 0.02 (SEM). Sample IO_7 did not align with the final pathological diagnosis by methylation classification and showed the classification of control inflammation with a calibrated score of 0.9. The result was presumably due to the low tumor percentage of 60% in the submitted specimen. Nevertheless, the CNV plot [51], in line with chromosomal microarray [64], accurately identified key chromosomal aberrations detected in glioblastomas, including chr 7 gain and chr 10 loss (Additional file 2: Fig. S9). Finally, the CNV plots, in accordance of clinically validated NGS, accurately detected all oncogene amplifications with both hg19 and T2T reference genomes [44, 56, 57] (n = 10/10, including EGFR, PDGFRA, CDK4, MDM4, MDM2, KIT) (Additional file 2: Fig. S9 and Additional file 3: Tables S7 and S8).
Distinguishing between astrocytoma, oligodendroglioma, and other lower-grade gliomas via conventional morphology-based intraoperative diagnosis is challenging to due to freeze artifacts. In addition, some glial neoplasms are defined by specific molecular alterations that could not be acquired during traditional intraoperative diagnosis. In our cohort, methylation profiling accurately classified astrocytoma, oligodendroglioma, and pleomorphic xanthoastrocytoma with a calibrated score of 0.99 ± 0.004 (mean ± SEM; n = 4). According to WHO classification, a required chromosomal aberration for oligodendroglioma diagnosis is 1p/19q codeletion which was observed in the oligodendroglioma (IO_6) by both our generated CNV plots [51] and chromosomal microarray [
留言 (0)