
記住我
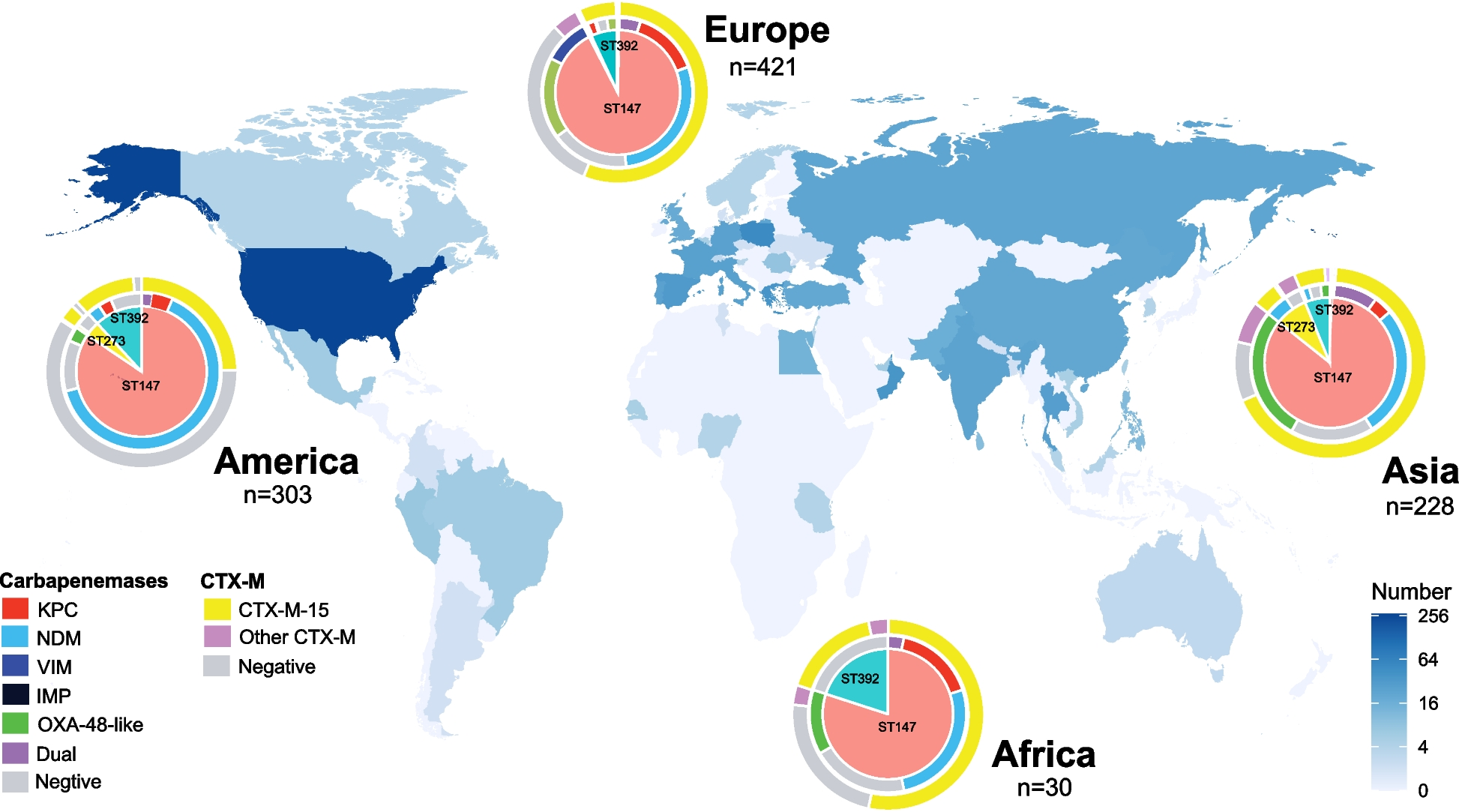








We compared the accuracy of our new tool, MixInfect2, against previous methods (MixInfect [6], SplitStrains [16], and QuantTB [18]) for detecting mixed infections and to estimate the major strain proportion from the dataset of 36 in vitro mixed samples and 12 non-mixed (“pure”) strains. The average coverage in all these samples was relatively high, ranging from 356- to 482-fold. We found that MixInfect2 accurately classified 36/36 mixed samples as combinations of two strains and all pure samples as single strains in the dataset (Fig. 1). In comparison, QuantTB identified four mixed samples as non-mixed (two at 90/10 and two at 95/5 mixing proportions) and two pure samples as mixes, as well as incorrectly predicting that six mixed samples were comprised of three strains. SplitStrains software correctly classified all mixed samples, but all pure samples were also predicted to be mixed strains, and MixInfect incorrectly predicted one 90/10 and eight 95/5 mixed samples as pure strains.
Fig. 1
The estimated major strain proportion against the known major strain proportion of the 36 in vitro mixed samples and 12 single strain samples was estimated using MixInfect2, MixInfect, SplitStrains, and QuantTB. Dashed red lines represent the true major strain proportion
Furthermore, the estimated major strain proportion of all in vitro mixed samples was close to the known major strain proportion using MixInfect2 (Fig. 1), and our new approach outperformed the other methods overall. In 70/30 major/minor strain proportion mixes, the median major strain proportion predicted by MixInfect2 was 69.0% (IQR 67.3–71.2), with the absolute difference between the predicted and known proportion significantly different to estimates from both QuantTB and SplitStrains (t-test P < 0.05). In 90/10 mixes, the median major strain proportion predicted by MixInfect2 was 89.2% (IQR 88.5–89.9), with the predictions significantly different to QuantTB and MixInfect (t-test P < 0.05). Finally, in 95/05 mixes, the median major strain proportion was estimated as 92.1% (IQR 91.8–92.3) and this was significantly different to QuantTB and SplitStrains (t-test P < 0.05).
Reconstructing constituent strain sequences of in vitro mixed samplesWe next compared three approaches for reconstructing both the major and minor constituent strain sequences of in vitro mixed samples as detailed in the “ Methods” section: (1) consensus allele frequency, (2) closest strain, and (3) closest strain + SNPs. For approaches that used a reference dataset to find the closest strain in non-mixed samples from the population, we included the sequences from a larger cohort of 2056 TB culture-positive individuals in the Karonga District of Malawi from which the constituent strains of the in vitro mixed samples were obtained. Of these isolates, 80 assemblies failed quality control, and 189 samples were identified as mixed infection using MixInfect2 and were removed, along with the 12 strains that matched the pure strains in the in vitro mixed dataset to avoid replication of these strains in the database. This resulted in a final reference database of 1775 non-mixed clinical strains and 12 pure strains from the in vitro dataset.
Reconstructing major strain sequencesFigure 2 shows the median SNP distance between the inferred major strain sequence and known constituent sequence for the 36 in vitro mixed samples predicted using the three approaches for reconstructing mixed sequences. We first tested the three approaches when the constituent strains of mixed samples (the pure strains from the in vitro dataset) were included in the reference dataset (Fig. 2A). The inferred sequence of the major strains was very close to the true sequence when estimated by all approaches (Fig. 2A), with all sequences predicted to be within 5 SNPs of the known sequence. The median SNP distance between the predicted sequence and true sequence was 0 SNPs for all methods, apart from the “closest strain + SNPs” method at 95% major strain proportion, which was 1 SNP, and there was no significant difference between the tested approaches (Kruskal–Wallis p > 0.05).
Fig. 2
Boxplots showing the SNP distance between the predicted and known major constituent strains of in vitro mixed samples using the three tested approaches. Boxes are colored by the different approaches used to predict the constituent strain sequence. Plot A shows the results when the known constituent sequences are included in the reference database, and plot B shows the results when these sequences are removed. Note that the vertical axis has been transformed by the square root for visualization
When the constituent strains of the mixed samples were not included in the reference dataset, the “closest strain” and “closest strain + SNPs” methods performed slightly worse than when consensus strains were included (Fig. 2B). Although there was still no significant difference among the tested methods when comparing the median SNP distance between the inferred and known sequences (Kruskal–Wallis p > 0.05), the average SNP distance was larger using these two methods when constituent sequences were removed from the database. Compared to the results when including the known constituent “pure” strain in the database, the median SNP distance between the inferred sequence and true constituent strain sequence increased to 1.5–2.5 SNPs, with a maximum of 136 SNPs difference. As the “consensus allele frequency” approach does not use a reference database, predicted sequences did not change with this approach when removing the constituent strains, resulting in the same median of 0 SNP distance and maximum of 5 SNP distance between predicted and known sequences. As such, the “consensus allele frequency” appears to offer the best approach to reconstruct the majority strain sequence of mixed infection.
Reconstructing minor strain sequencesThe performance of the tested approaches for reconstructing the minor strain sequences of the in vitro mixed samples was impacted more by the proportion of the minor strain in the mix than for the major constituent strain. At the 30% minor strain proportion, the “closest strain” method inferred minor strain sequences that most closely matched known constituent sequences, with a median of 0 SNPs and a maximum of 3 SNPs difference (Fig. 3A). The “closest strain + SNPs” method also found closely linked sequences with a median 0 SNP distance between predicted and known sequences but with a higher maximum distance of 7 SNPs. The “consensus allele frequency” method performed significantly worse (Kruskal–Wallis p < 0.05), with a median of 5.5 SNPs between the predicted and known constituent sequences. Removing the true constituent sequences from the dataset when searching for the closest strains increased the median SNP distance of the “closest strain” and “closest strain + SNPs” methods to 2 SNPs (Fig. 3B). This was still lower than the “consensus allele frequency” approach but the difference between the tested methods was now not significant (Kruskal–Wallis p > 0.05).
Fig. 3
Boxplots showing the SNP distance between the predicted and known minor constituent strains of in vitro mixed samples using the three tested approaches. Boxes are colored by the different approaches used to predict the constituent strains. Plot A shows the results when the known constituent strains are included in the list of “pure” strains, and plot B shows the results when these strains are removed. Note that the Y-axis has been transformed by the square root for visualization
When the minor strain proportion was at 10% in the in vitro mixed samples, the “closest strain” approach performed significantly better than other methods (Kruskal–Wallis p < 0.05), with a median SNP distance between the predicted and known constituent sequence of 0 SNPs. This is compared to 131.5 SNPs using the “consensus allele frequency” method and 93.5 SNPs using the “closest strain + SNPs” method (Fig. 3A). While there was one outlier sequence with a large SNP distance between the predicted and true sequence using the “closest strain” approach (maximum 187 SNPs), all other sequences were predicted within 3 SNPs of the true sequence. In addition, when the known constituent sequence was removed from the dataset, this method still outperformed the other approaches significantly and the median SNP distance between predicted and true sequences was 2 SNPs (Fig. 3B).
At a 5% minor strain proportion in mixed samples, the median SNP distance between the predicted sequence and known constituent strain sequence was high using all tested approaches (Fig. 3). Inspection of the VCF file showed that many sites that differed between the minor and major constituent strains in the 95/5 samples were called as a cSNP matching only the allele of the major strain sequence instead of hSNPs. This was due to the very low number or absence of reads carrying the allele from minor constituent strain. This was also evidenced by the large difference in the SNP distance between the closest strain identified in the reference database and the known constituent strain in most of the in vitro minor strains at a 5% mixing proportion (Additional File 1: Fig. S1). In these samples, the closest strain identified was often very divergent from the known constituent strain and in some instances was closer to the major constituent strain, which was a different MTBC major lineage in some of the mixed samples. Thus, it appears that a minor strain proportion of 5% is too low to accurately infer the minor strain sequence using the WGS data and approaches considered here.
Sensitivity analysisTo assess how the size and composition of the dataset affected the performance “closest strain” and “closest strain + SNPs” methods, we downsampled the dataset of “pure” isolates in the Karonga dataset to 50% and 75% of the original size and re-calculated the SNP distance between the predicted and known major and minor sequences for all in vitro mixed samples. This process was repeated 100 times at 50% and 75%, randomly selecting “pure” strain sequences to include in the new dataset. We found that the median SNP distance of the “closest strain” method increased from 0 SNPs at all major strain proportions to 3 SNPs with a 50% downsampled dataset (Additional File 1: Fig. S2A) and 4 SNPs with the 75% downsampled dataset (Additional File 1: Fig. S2C). The median SNP distance between predicted and known major strain sequences increased from 0 SNPs using the “closest strain + SNP” approach to 2–5 SNPs in the 50% and 4–5 SNPs in the 75% downsampled dataset. Furthermore, the maximum distance to the true sequences when using these approaches increased dramatically in the downsampled datasets where sequences that were closely related to the major strain in the mixed sample may not have been included. As such, using the “consensus allele frequency” approach appears to be the best option to predict major constituent strain sequences to mitigate the possibility that close “pure” strains are not included in the dataset.
For the minor strain sequence prediction, the optimal method to reconstruct the sequence appeared to depend more on the completeness of sampling and sequencing of the infected population. While the “closest strain” approach performed the best in the 30% minor strain proportion samples with the full dataset, when the dataset is downsampled by 50% and 75%, “consensus allele frequency” achieves only a slightly higher median SNP distance between predicted and known sequences than the other approaches (5.5 SNPs compared to 2–4 SNPs) but with far fewer samples with a predicted sequence a large distance from the true constituent sequence (Additional File 1: Fig. S2B and S2D). At 10% minor strain proportion, the “closest strain” approach still performs best for estimating the minor strain in both the 50% and 75% downsampled datasets, but again, the number of predicted sequences that were a large SNP distance to the known sequence was high. Therefore, in populations where sampling or sequencing is sparse and the likelihood of including the constituent strain or a closely related sequence is low, it may be problematic to predict minor strain sequences, particularly for low minor strain proportions. In this instance, it may be optimal to use the “consensus allele frequency” method to predict both major and minor constituent sequences but not attempt to reconstruct minor strains at low mixing proportions to reduce the chances of poorly predicting the minor strain sequence.
In our bioinformatic pipeline, hSNPs were defined as sites with more than one allele supported in aligned reads and a minimum minor allele depth of 10 reads. We assessed the impact of characterizing hSNPs in this way by comparing the SNP distance between the predicted sequence and the true constituent strain sequence using the “closest strain” approach when changing the metrics used to call hSNPs. These included lowering the minimum minor allele read depth to 5 reads, using a minimum allele read proportion (rather than raw depth) of 0.01, 0.02, 0.05 and 0.1 and using the heterogeneous base call (e.g., “0/1”) by setting the ploidy option the variant-calling software to diploid. We found no significant change in the results of reconstructing the major and minor constituent strains of the in vitro mixes using the “closest strain” approach using the different hSNPs calling methods, apart from an increase in the distance between the predicted and known sequence at the 10% minor strain proportion when using a minimum minor allele proportion of 0.1 (Additional File 1: Fig. S3).
Mixed TB infection and transmission in MoldovaWe used a real-world dataset of 2220 Mtb isolates from the Republic of Moldova [21] to identify the proportion of mixed infection in this population and reconstruct the constituent strain sequences. A total of 146 of 2220 (6.6%) isolates were identified as mixed infection using MixInfect2 (Additional File 2: Table S1), substantially fewer than previously predicted in this dataset using the earlier MixInfect approach (386/2220; 17.4%) [21]. All major constituent strain sequences were predicted using the “consensus allele frequency” approach as this achieved the best results in the in vitro mixed samples. The “closest strain” approach was used to predict minor strain sequences with an estimated minor strain proportion of ≥ 10%. With this approach, we also set a maximum distance threshold of 1000 SNPs to the closest strain in the reference database to reduce the chance of matching a strain that is very divergent to the true constituent strain; if the closest strain was further, then no minor strain sequence was predicted. We did not reconstruct the major or minor strain in mixed samples with a major strain proportion estimate of ≥ 50% and < 60% (N = 15) as we have not reliably tested these methods when constituent strain proportions were close to parity. Eleven isolates with a high proportion of hSNPs (≥ 10% of total SNPs) that were not classified as mixed infection were flagged and removed from further analysis. This removal resulted in a final dataset of 2291 isolates: 2063 “pure” strain, 129 major strain, and 99 minor strain sequences.
In the 99 mixed samples for which both the major and minor strain sequence was predicted, we found that 45 (45.4%) contained a mix between different major lineages and a further 23 (23.2%) were mixes of lineage 4 sub-lineages (all lineage 2 strains were of the Beijing lineage 2.2). We also found evidence of hetero-resistance to isoniazid and/or rifampin in 27/99 mixed infections (27.3%), and while this can occur in single-strain infections when these SNPs can be under selection and not reached fixation, this proportion was higher than hetero-resistance found in 20/2063 non-mixed strains (Chi-square test 295.08, P < 0.05). A maximum likelihood phylogeny that included both non-mixed and the predicted constituent mixed strain sequences showed that most mixed constituent strains were closely related to other sequences in the dataset, although there were a small number of major strains that appeared relatively genetically distant to any other strain in the dataset (Additional File 1: Fig. S4).
Finally, transmission clusters were constructed by linking all sequences that were separated by a pairwise SNP distance of ≤ 5 SNPs, including the predicted major and minor mixed constituent strains. We identified 90 clusters that contained at least one constituent strain of the mixed infections, including a large transmission cluster containing 130 sequences with six major constituent and nine minor constituent strains of mixed infections and a large cluster of 66 sequences containing two major constituent and one minor constituent (Fig. 4). A total of 45 of 129 (34.9%) major constituent strains were predicted to be part of a transmission cluster compared to 951/2063 (46.1%) of pure strains (Chi-square test 5.71, P = 0.02). A total of 96 of 99 minor constituent strains were found in transmission clusters, although this high number of minor strains included in clusters was due to the “closest strain” method predicting the minor constituent sequence to be nearly identical to the sequence of the closest “pure” strain in most instances. There also appeared to be one cluster comprising only one non-mixed strain that was the closest strain sequence to 14 minor strains. While it is possible that this strain is closely related to the minor strain in these mixed samples, this could also be explained by cross-contamination in these samples rather than clinical mixed infection. The full list of cluster designations with the number of sequences and constituent sequences of mixed infections can be found in Additional File 2: Table S2.
Fig. 4
Visualization of the eight largest transmission clusters (N > 10) in the Moldova Mtb dataset that contained at least one mixed constituent strain produced using TGV (https://jodyphelan.github.io/tgv/) [32]. The color of the node represents whether the sample is non-mixed (green), the major constituent strain of mixed infection (blue), or minor constituent strain of mixed infection (red). Edges represent any pairwise distance between sequences of ≤ 5 SNPs and the node shape denotes the MTBC major lineage, where circle nodes are lineage 4 strains and triangle nodes lineage 2 strains
留言 (0)