
記住我





The catalytic activity of ALDH2 is NAD+-dependent; hence, binding of the coenzyme is essential for catalytic activity [17]. Molecular docking is a powerful tool and widely used method in molecular modeling. It involves predicting the preferred orientation of a molecule when binding to a target protein [36]. In the context of ALDH2 variants, docking can reveal how mutations affect coenzyme binding, which is crucial for understanding their impact on enzyme activity. At this point, NAD+ was docked to the prosthetic site of wild type and mutants ALDH2 using AutoDock4.2. The binding energy score and inhibition constant were used to evaluate the strength of protein–ligand interactions and rank them. A more negative binding energy score indicates a more favorable binding between the ligand and the protein.
The docking process of NAD+ onto the prosthetic site of ALDH2 shows that the wild type exhibits the highest binding affinity at the lowest concentration. Among the natural variants, only E487K significantly lowers binding affinity and has a higher inhibition constant (Table 1). This suggests that this mutant significantly impacts the interaction between NAD+ and ALDH2, potentially affecting the enzyme’s activity and function.
Table 1 Binding energies and inhibition constants of NAD against Wild type and mutabtsDocking can provide information on how a mutation affects ligand binding with its target at the molecular level based on visual inspection of the interaction. Accordingly, using the Discovery Studio visualizer and PyMol software, the interactions of wild type and mutants with NAD+ were explored in depth by analyzing their binding patterns. A comparison of the docked poses of NAD+ in wild type (cocrystallized) and mutants ALDH2 showed a similar interaction pattern (Fig. 2). The formation of a similar binding pattern confirmed that the docking simulation study was reliable for reproducing the experimental binding mode of ALDH2. In the binding pose of the docked structures, hydrogen bond interactions were observed. In addition, dominant electrostatic and vdW interactions are also present.
Fig. 2
Pose view and protein–ligand interactions of the wild type and mutant structures
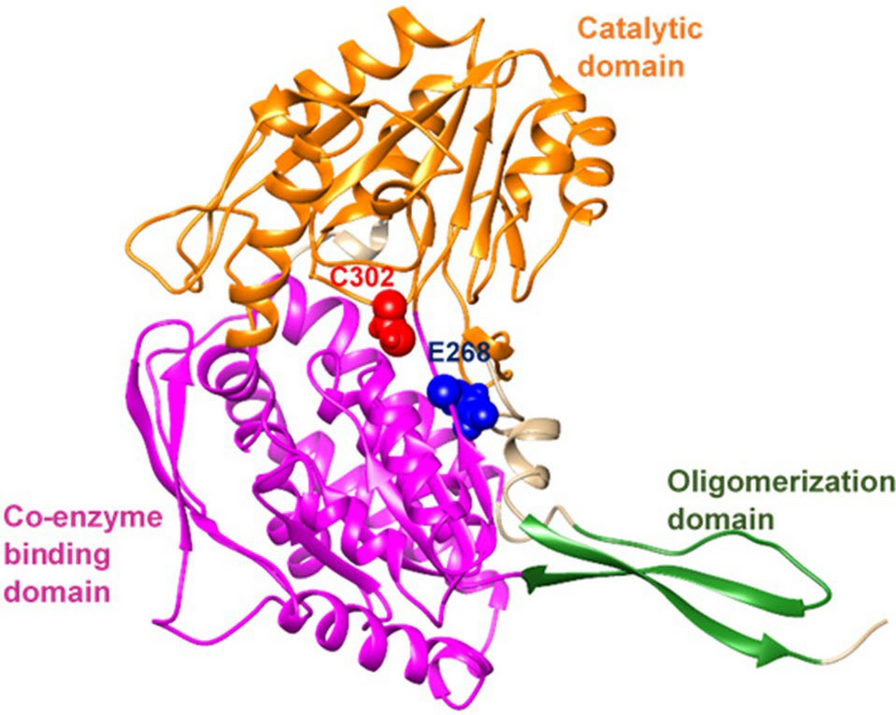
In the wild type, eight residues Gln349, Leu269, Gly245, Glu268, Trp168, Ile166, Glu195, and Lys192 formed single hydrogen bond with NAD+ at prosthetic site. Several residues formed other interactions such as hydrophobic residues of Phe401, Pro167, Ilu165, and Pro226 (Pi-Alkyl), Phe243 and Gly225 (Pi-Pi Stacked), acidic, negatively charged Asp346 (Unfavorable Donor-Donor), and Ile249 (Pi-Sigma). NAD+ docked into the prostatic site of E320V and E479K phenotypes with a docking score close to the wild type, −8.86, −8.99, and −9.09 kcal/mol, respectively (Table 1). At the docking pose of each of the E320V and E479K phenotypes, seven hydrogen bonds are observed. Analyzing the binding amino acid residues reveals that in addition to some interactions such as Pi-Alkyl, Pi-Pi Stacked, and Carbon H-Bond, they form 14 and 8 van der Waals (Vdw) interactions, respectively. However, the computational docking suggests that the substitution of negative glutamic acid (E) with positive lysine (K) at position 487 could lead to a decrease in the hydrogen bonding network and the charge distribution in the ALDH2. For example, the total interactions decreased from 20 in the wild type to 16 in E487K, which subsequently caused a significant reduction in binding energy from −9.09 to −8.16 kcal/mol. Interestingly, the hydrogen bonds decreased significantly, from 8 in the wild type to 5 in E487K. Also, the binding pose shows that the lysine substitution at position 487 disrupts any interaction of NAD+ with Cys302 and Glu268 which is critical for the catalytic activity of the enzyme. In the wild type, a hydrogen bond and vdW interaction is seen between Glu 268 and NAD+, Cys302 and NAD+, respectively (Fig. 2). In Table 2, the binding interactions of NAD+ against the prostatic site of wild type and mutants of ALDH2 are summarized.
Table 2 Binding interactions of NAD+ against the prostatic site of wild type and mutants of ALDH2 Substitution effects on structure via molecular dynamics simulationsA close and intricate relationship exists between protein stability, function, and activity. This relationship is influenced by the protein’s response to amino acid substitution, which can lead to significant changes in stability. Missense mutations often disrupt the folding of polypeptides into their functional conformations, leading to increased instability and contributing to disease [37].
We conducted 100 ns molecular dynamics simulations to investigate the potential deleterious effects of selected mutations on the protein’s structural dynamics. Molecular dynamic simulations show how mutations impact protein conformation, stability, and function. These simulations provide intricate atomic-level details, facilitating comprehension of disease mechanisms. The RMSD estimates the deviation in the backbone atoms of the protein residues from initial structural conformation to final position, indicative of the degree of protein stability. A high RMSD indicates increased mobility of the C-α atom indicative of structural instability, which could correlate with its dysfunctionality, while low RMSD value corresponds with minimal motions in the backbone atoms and protein stability.
The RMSD values of wild type and mutants of ALDH2 throughout the MD simulations are shown in Fig. 3a. The wild type, E320V, and E479K phenotypes show similar deviations from their starting structures till the end of the trajectory. In the case of the E487K mutant, the RMSD value was consistently higher during most of the 100 ns simulation, particularly at the end of 40 ns. This suggests that this mutant probably leads to significant protein conformational changes and destabilizes the protein structure compared to the wild type.
Fig. 3
a Root mean square deviation (RMSD); b root mean squared fluctuation (RMSF); and c radius of gyration (Rg); profiles of the wild type and mutants ALDH2 during MD simulation
To compare the impact of mutation on the residual fluctuation, the root mean squared fluctuation (RMSF) values were calculated for the C-α backbone atom at each time point within the trajectories of both the native and mutant structures. An elevated RMSF value signifies greater flexibility, whereas a reduced value suggests diminished flexibility and structural rigidity. Dysfunctionality in enzyme activity may be result of these alterations. The data presented in Fig. 3b indicates that the fluctuations of the E320V and E479K ALDH2 variants are close to those of the wild type. In contrast, the E487K mutant demonstrated higher RMSF value in specific regions, indicating localized flexibility. Notably, this variant displayed increased flexibility in three specific regions: (135–150), (245–260), and (485–490), which correspond to the coenzyme binding domain, catalytic domain, and oligomerization domain, respectively. This suggests that while the overall structure may not fluctuate significantly, certain areas of the protein experience increased movement. Such localized flexibility could contribute to the destabilization of the protein, potentially affecting its function.
The radius of gyration (Rg) is a useful parameter for measuring the overall dimension and compactness. A high Rg value suggests a greater number of residues exposed to the solvent, which could be an indicator of protein instability. Accordingly, the Rg plot in Fig. 3c revealed that the E487K mutant induced increased Rg and then less compactness, while it was lower and similar trends of Rg trajectory and compactness in the wild type, E320V, and E479K forms. Hence, in the case of the E498K mutant, distortion of structural integrity by residual configurational perturbations that can cause enzyme dysfunction is expected.
Evaluation of the functional impact of coding nsSNPsTo get more accurate results, selected missense nsSNPs were analyzed by four in silico tools—SIFT, PolyPhen-2, SNAP, and PredictSNP—to assess potentially deleterious missense nsSNPs. Protein sequence and amino acid residue variations with the corresponding mutational positions were input into these servers. The findings are presented in Table 3. The tools predicted deleterious missense nsSNPs based on their default scoring parameters. Among the three natural variants examined, only E504K (E489K) was identified by all four bioinformatics tools as potentially deleterious and likely to impact protein function. The data presented in Table 2 indicate a notable correlation between the outcomes derived from bioinformatics tools based on sequence information and those obtained from structural-based molecular dynamics simulations for the three predicted nsSNPs. This finding implies that the E489K nsSNP is the sole variant that adversely affects both the function and structure of the protein.
Table 3 The prediction results of natural nsSNPs of human ALDH2 using SIFT, PolyPhen-2, SNAP, and PredictSNP algorithms
留言 (0)