
記住我
Recent advancements in cancer treatment include the utilization of replication-competent viruses for selective oncolysis of tumors, known as oncolytic viruses (OVs), while sparing normal cells. This therapeutic approach can be employed either as a standalone treatment or in combination with other therapies to inhibit tumor progression (1). OVs encompass both wild-type and genetically modified variants. Genetic engineering strategies aimed at modifying these viruses involve deleting specific genes to limit toxicity to healthy cells (2, 3), inserting genes to activate the immune system, stimulate immune responses, or inhibit angiogenesis (4–6), and combinations of these strategies.
Herpes simplex virus type 1 (HSV-1) possesses a genome consisting of 152 kb of double-stranded linear DNA that encodes approximately 85 protein-coding genes, with 47 being dispensable in cell culture (7). HSV-1 has several advantages that position it as a leading candidate for oncolytic virotherapy (OVT). Notably, its genome contains two unique segments: one is the unique long (UL) segment and the other is the unique short (US) segment; each is flanked by inverted repeat (IR) elements. This genomic architecture allows for the insertion of fragments exceeding 30 kb or deletion of multiple virulence genes without compromising its lytic replication cycle within tumor cells (8, 9). Additionally, anti-herpetic drugs can inhibit HSV-1 replication in cases of accidental infection (10). Importantly, HSV-1 does not integrate into the host genome nor induce insertional mutations (7). Due to the above characteristics of HSV-1 virus, it has three advantages compared to other oncolytic viruses. First, it has a larger genome that can insert and accommodate multiple foreign genes. Additionally, the use of acyclovir can easily control HSV-1 infections in non-tumor cells. Finally, theoretically, HSV-1 has a lower likelihood of causing insertional mutagenesis in infected cells.
Currently, there are two primary directions for genetic modification of oncolytic HSV-1 (Figure 1). The first direction focuses on reducing HSV-1’s infectivity and toxicity towards normal cells by limiting viral replication and assembly (3, 11–13), modifying proteins that bind viral receptors (14), and decreasing mechanisms involved in viral immune evasion (15, 16). The second direction aims to enhance HSV-1’s anticancer efficacy through interference with tumor cell metabolism (17), induction of autophagy (18, 19), improvement in immune recognition processes (20–23), and alteration of the tumor microenvironment itself (6, 24, 25). Several key gene modifications related to silencing specific genes or introducing exogenous genes into the HSV-1 genome will be discussed separately.
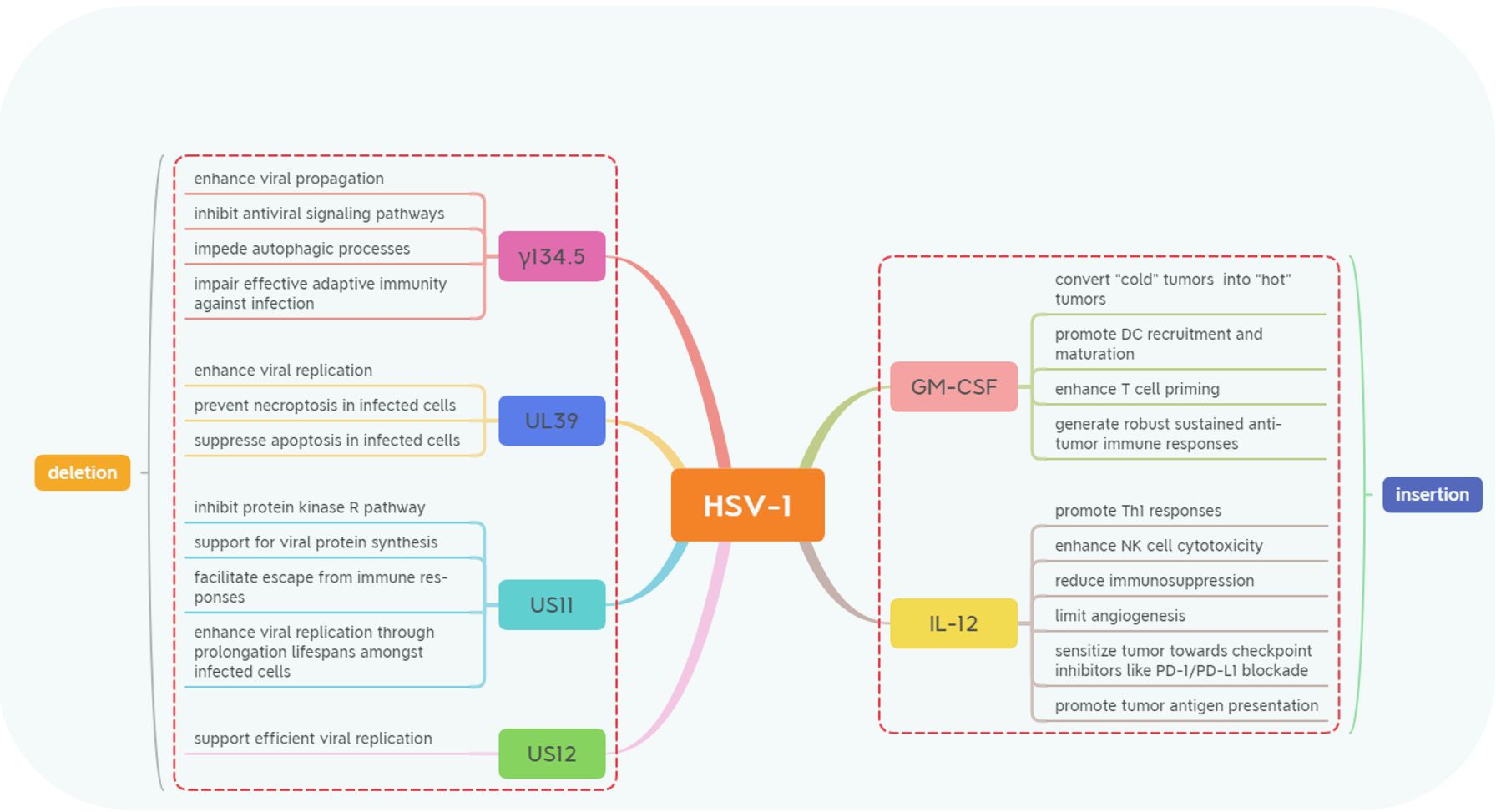
Figure 1. The genetic modifications in Herpes Simplex Virus-1 (HSV-1) involve deletions and insertions. These majority modifications include the deletion of genes such as γ134.5, US11, US12, and UL39 and the expression of transgenes like GM-CSF and IL-12. These strategic genetic engineering techniques are designed to enhance the oncolytic properties of HSV-1 while modulating immune responses to improve anti-tumor efficacy through various mechanisms.
2 Silencing genes2.1 Gene γ134.5To enhance the selectivity of HSV-1 for infecting epithelial-derived malignancies while minimizing the risk of infection in healthy somatic cells and preventing uncontrolled spread of HSV-1 to normal somatic cells, numerous research groups knockout the γ134.5 gene. The diploid gene γ134.5 located within the inverted terminal repeats flanking the long unique sequence of HSV-1 DNA is classified as a gamma-late or “leaky late” gene. It encodes ICP34.5, a neurovirulent protein. ICP34.5 consists of 263 amino acids organized into three main domains: the N-terminal domain, the linker region, and the C-terminal domain. Those domains are responsible for binding host proteins essential for both viral replication and immune evasion (26–29).
The principal function attributed to ICP34.5 involves enhancing viral propagation across peripheral tissues alongside central nervous systems, contributing significantly toward HSV, and inducing neurovirulence via various mechanisms including protein phosphatase I (PPI) dephosphorylating eIF2α, thus preventing shutoff from host protein synthesis while enabling continuous production (11, 30, 31). Furthermore, ICP34.5 converts proliferating cell nuclear antigen (PCNA) from repair mode back toward a replicative state, crucially initiating HSV replication (32).
Moreover, ICP34.5 inhibits antiviral signaling pathways ensuring persistent infections. ICP34.5 disrupts retinoic-acid-inducible gene I (RIG-I) signaling preventing interaction between RIG-I and mitochondrial antiviral signaling protein (MAVS), a pivotal adaptor inhibiting downstream activation IRF3 and subsequent IFN production (33). Stimulator interferon genes (STING) is another important player during antiviral responses where N-terminal domain binds/inactivates STING, thereby diminishing IRF3 activation/IFN secretion (34).
Additionally, ICP34.5 impedes autophagic processes through Beclin binding interactions specifically targeting Beclin-1 (Atg6) (35). Such engagement hinders this vital cellular defense mechanism allowing enhanced pathogenesis while blocking class II antigen presentation, further augmenting HSV virulence (36, 37).
Lastly, ICP34.5 also disrupts NF-kB activation suppressing dendritic maturation and ultimately impairing effective adaptive immunity against infection. The N-terminal domain of ICP34.5 interacts with IKKα/β, components of the IκB kinase complex, while its C-terminal domain recruits PP1α. This interaction leads to the dephosphorylation of IκB kinase, preventing the activation of NF-κB, a transcription factor that regulates genes involved in immune responses, inflammation, and cell survival (38, 39).
Through these combined mechanisms, ICP34.5 serves as a critical factor in HSV-1 pathogenesis by supporting viral replication, evading multiple immune pathways, and altering host cellular functions. However, after silent gene γ134.5, the oncolytic efficacy of HSV-1 in malignant tumors of neurological origin (e.g., glioblastoma and neurofibroma) has decreased. In clinical treatment, it is necessary to choose the oncolytic HSV-1 with silent gene γ134.5 according to the tissue source of the tumor.
2.2 Gene US11To reduce immune evasion and subsequent uncontrolled viral infection after the injection of oncolytic HSV-1 into patients, gene US11 was selectively silenced. It can not only reduce the immune evasion of oncolytic HSV virus immunocompromised patients but also decrease the replication and spread of the virus in healthy cells. The US11 protein is a small basic phosphoprotein with a molecular weight of approximately 18 kDa. Its coding region extends from the ATG codon at residue 12,641 to the TAG stop codon at residue 12,158, resulting in a protein mass of 17,756 Da. The carboxy-terminal half contains several arginine-X-proline (R-X-P) repeats that confer RNA-binding capability (40). These repeats also harbor nucleolar import and nuclear export signals that facilitate localization within both nucleus and cytoplasm as required. Encoded by the late γ2 gene, US11 is expressed during later stages of HSV infection and performs several crucial functions enhancing HSV-1 survival within host cells (41).
Inhibition of protein kinase R (PKR) pathway alongside support for viral protein synthesis are primary functions attributed to US11. The PKR pathway becomes activated upon binding double-stranded RNA (dsRNA), leading to phosphorylation of eIF2α, an event typically halting protein synthesis as part of an antiviral response. US11 exhibits high affinity for dsRNA, allowing it to sequester this molecule away from PKR (42, 43). By obstructing PKR activation through this mechanism, US11 prevents eIF2α phosphorylation, thus sustaining viral protein synthesis. Furthermore, when expressed early during infection, US11 can partially compensate for ICP34.5’s function inhibiting eIF2α phosphorylation. This redundancy enables HSV-1 to maintain ongoing translation even if ICP34.5 is absent. However, both proteins are generally necessary for full resistance against type I interferon (IFN) responses (43).
Additionally, US11 modulates various host antiviral pathways facilitating escape from immune responses by HSV-1. During late phases when levels of dsRNA peak, US11 binds/sequesters dsRNA effectively preventing MDA5/RIG-I activations, which subsequently suppress IRF3 activity along with interferons production (44, 45). Such inhibition impedes induction of ISGs establishment, hence compromising antiviral states among infected cells. Another important mechanism involves oligoadenylate synthetase (OAS) pathway activated via dsRNA binding where US11 inhibits OAS activity, blocking RNase L, thereby aiding virus evade degradation while preserving infectivity (46).
Moreover, US11 plays pivotal roles regulating cell survival pathways ultimately promoting enhanced replication through prolongation of lifespans among infected hosts. Within nuclei, US11 interacts with homeodomain-interacting protein kinase HIPK2 responding stress signals including those arising from ER-regulating cycle progression/pro-apoptotic signaling (47). By antagonizing growth-arrest-induced HIPK2, HSV-1-infected cells evade apoptosis, continuing to facilitate virion propagation (48).
Through multifaceted functionalities, US11 facilitates HSV-1 replication by preventing translational shutoff, inhibiting immunological signaling and obstructing pro-apoptotic response. By suppressing activations across PKR, OAS, MDA5, and RIG-I enable HSV-1 to evade defenses and sustain syntheses, thus augmenting survivability and pathogenicity. After the silencing of US11, the therapeutic effect of a single injection of oncolytic HSV-1 may be transient. Throughout the course of treating malignant tumors, multiple injections of oncolytic HSV-1 are required, and continuous monitoring of tumor growth is necessary to evaluate whether to administer oncolytic HSV-1 again.
2.3 Gene US12One of the immune evasion mechanisms of HSV-1 is to inhibit antigen presentation by binding to TAP, thereby preventing cytotoxic T cells from recognizing infected cells. The protein encoded by the US12 gene is key to binding with TAP. The US12 gene (ICP47) spans residues 12,972 to 12,708 and encodes the immediate-early protein ICP47, which consists of 88 amino acids. Similar to US1 at the opposite end of the Us region, both the promoter region of ICP47 and a significant portion of its 5′-non-coding mRNA are situated within the terminal repeat (TR) sequences of HSV-1 (26). ICP47 plays a pivotal role in HSV-1’s immune evasion strategy through various mechanisms and polymorphic functions during different stages of infection. It exhibits high-affinity binding to the transporter associated with antigen presentation (TAP). By occupying TAP’s substrate-binding site, ICP47 inhibits viral peptide loading onto MHC class I molecules for presentation on cell surfaces to CD8+ T cells, effectively blocking cytotoxic T lymphocyte (CTL) recognition of infected cells and enabling HSV-1 to evade immune detection (14, 49).
The function of ICP47 is polymorphic as infection progresses. During early infection stages, it may impede RNA splicing, thus limiting host and viral gene expression in a tightly regulated manner. In later stages, however, ICP47 appears to facilitate viral mRNA export from the nucleus into the cytoplasm, thereby supporting efficient viral replication (50).
Deletion of US12 has been shown to enhance HSV-1’s oncolytic potential and tumor-cell-killing ability alongside a stronger immune response. This deletion places US11 under immediate-early promoter control while enhancing replication efficiency in tumor cells for HSV-1 strains lacking ICP34.5, suggesting promising applications for oncolytic virotherapy (51, 52).
In summary, when ICP47 is deleted from its genome context, it can serve as an effective tool to reduce immune evasion in immunodeficient environments and tumor cells.
2.4 Gene UL39The UL39 gene is situated within the Unique Long Region of the HSV-1 genome and plays a critical role in viral replication and in modulating physiological processes within host cells (53). To reduce the spread of oncolytic HSV-1 proliferation within tumors, gene UL39 is selected as the candidate gene to be silenced. Unlike certain other HSV-1 genes, UL39 does not generate repetitive sequences with adjacent regions of the viral genome, rendering it structurally distinct. This gene is expressed early during the HSV-1 replication cycle, prior to the entry of the viral genome into the host cell nucleus (54, 55). Its initial translation depends on transcription and translation mechanisms within host cells, enabling HSV-1 to swiftly produce essential proteins for sustained infection (55).
ICP6, which is encoded by the UL39 gene and serves as the large subunit of ribonucleotide reductase, is vital for converting ribonucleotides into deoxyribonucleotides necessary for DNA synthesis in viruses (56). Additionally, ICP6 can phosphorylate eIF2α, a key initiation factor, thereby suppressing host protein synthesis and favoring production of viral proteins over cellular functions. This mechanism facilitates enhanced viral replication within infected hosts (57).
Another significant function attributed to ICP6 involves its modulation of programmed cell death (PCD) processes in infected cells through its receptor-interacting protein-homotypic interaction motif (RHIM) (54). The RHIM domain prevents necroptosis by obstructing RIPK1-RIPK3 complex formation (receptor-interacting protein kinases 1 and 3) in human cells (58). Furthermore, it promotes aggregation of RIPK1 that subsequently undergoes degradation via aggrephagy, further diminishing necroptotic activity (11). It also inhibits RIPK1/RIPK3-dependent necroptosis in human cells. Beyond preventing necroptosis, ICP6 additionally suppresses apoptosis by directly binding to and inhibiting caspase-8. This dual inhibition strategy allows HSV-1 to circumvent major apoptotic pathways while promoting both survival and proliferation within host environments (53).
Inactivation of ICP6 through fusion with LacZ results in restricted virus propagation primarily among dividing cells, particularly tumor cells capable of supplying deoxyribonucleotides via endogenous pathways (59). This tumor-specific characteristic exhibited by mutated forms of ICP6 positions HSV-1 variants makes them promising candidates for oncolytic therapies. Although silencing the gene UL39 can limit the proliferation of oncolytic HSV-1 after injection, enhancing the safety of this oncolytic virus in clinical applications, the tumor-killing effect of this oncolytic virus is also restricted, requiring a larger dosage and multiple injections to achieve the desired effect.
3 Inserting exogenous genes3.1 GM-CSFInduction of immune cells to kill tumor cells is one of the key mechanisms of oncolytic virus anticancer. In addition to the immune activation of the viral particles themselves, the cytokine genes carried by oncolytic viruses can be synthesized and released in tumor cells, and this process also significantly improves the killing efficacy of immune cells to tumor cells. Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a multifunctional cytokine that plays critical roles in immune modulation, serving as a bridge between hematopoiesis and immune activation (60, 61). Initially identified as a growth factor that stimulates the differentiation of bone marrow progenitor cells into granulocytes and macrophages, GM-CSF also activates various signaling pathways, including JAK/STAT, MAPK, and PI3K, through JAK2 activation, thereby influencing immune functions (62–65).
GM-CSF enhances the survival, proliferation, and differentiation of myeloid lineage cells such as neutrophils, macrophages, and dendritic cells (DCs) (66). By promoting DC maturation, GM-CSF improves antigen presentation capabilities and T-cell activation (60). To enhance the phagocytic abilities of macrophages and their anti-tumor activities, GM-CSF drives the polarization of these cells from an M2 (anti-inflammatory) phenotype to an M1 (pro-inflammatory) phenotype (67). Furthermore, GM-CSF strengthens immune recognition of cancer cell neoantigens by fostering antigen, presenting cell generation, and elevating major histocompatibility complex (MHC) expression, thereby reinforcing the overall immune response against tumors (23, 68).
Oncolytic viruses (OVs) armed with GM-CSF lead to localized cytokine expression within the tumor microenvironment while enhancing tumor cell susceptibility to viral infection by driving these cells into the cell cycle. This effectively converts “cold” tumors characterized by low immune activity into “hot” tumors exhibiting high levels of immune activity (69). Additionally, GM-CSF-armed OVs promote DC recruitment and maturation at tumor sites, which enhances T-cell priming and generates robust anti-tumor immune responses (70). This process can also foster long-term immunological memory resulting in sustained anti-tumor effects. The first oncolytic HSV-1 armed with GM-CSF, talimogene laherparepvec (T-VEC), demonstrated significant anti-tumor efficacy leading to FDA and EMA approval for melanoma therapy (71–73). However, excessive GM-CSF release also aggravates the systemic symptoms, such as fatigue and elevated body temperature. More attention is paid to the inflammatory status of patients during treatment.
3.2 IL-12To enhance the tumor resistance of NK cells and cytotoxic T lymphocytes, interleukin-12 (IL-12) was selected as a candidate gene for the insertion of oncolytic HSV-1. Its ability to reshape the tumor microenvironment while augmenting responses to checkpoint inhibitors underscores its therapeutic potential particularly when combined with other cancer immunotherapies establishing it as a formidable agent in anti-tumor immunity.
IL-12 facilitates CD4+ T-cell differentiation into Th1 cells that secrete elevated levels of interferon-gamma (IFN-γ), which subsequently activates NK cells and cytotoxic T lymphocytes (CTLs), thereby enhancing their anti-tumoral functions (74). Moreover IL-12 amplifies both growth rates and cytotoxic activities among NK cells alongside CD4+ and CD8+ T lymphocytes, resulting in increased production of perforin and granzyme B, which are key molecules essential for CTLs’ capacity to eradicate tumor cells (74, 75). Additionally, IL-12 promotes differentiation toward memory or effector T-cell phenotypes, thus improving precision persistence within targeting residual or metastatic malignant populations (76, 77).
Furthermore, IL-12 diminishes regulatory T-cell (Treg) and myeloid-derived suppressor cell (MDSC) populations within tumoral environments alleviating suppression mechanisms detrimental toward effective antitumoral responses (5, 78). It also drives macrophage polarization toward an M1 phenotype, a state characterized by pro-inflammatory properties conducive for inducing tumoricidal activity. By downregulating vascular endothelial growth factor (VEGF), IL-12 effectively reduces angiogenesis associated with tumors (25, 79).
Moreover, IL-12 sensitizes neoplasms toward checkpoint inhibitors like PD-1/PD-L1 blockade, thereby amplifying therapeutic efficacy (24, 80). With regard to promotion of tumor antigen presentation, death induced through IL-12-stimulated effectors releases TAAs further stimulating adaptive immunity assisting remaining malignant targets recognized by activated T cells (76, 77).
4 OthersIn addition to the aforementioned wide-ranging applications in genetic modification techniques, several strategies for genetic modification demonstrate significant potential for clinical application (Table 1). A category of genetically modified viruses has been developed to enhance viral replication and tumor-specific cytotoxicity. The IRS1 and TRS1 genes from human cytomegalovirus (HCMV) have been inserted into HSV-2 to improve protein synthesis and replication by inhibiting PKR kinase activity and autophagy, thereby facilitating robust viral protein production and survival within tumor cells (81).
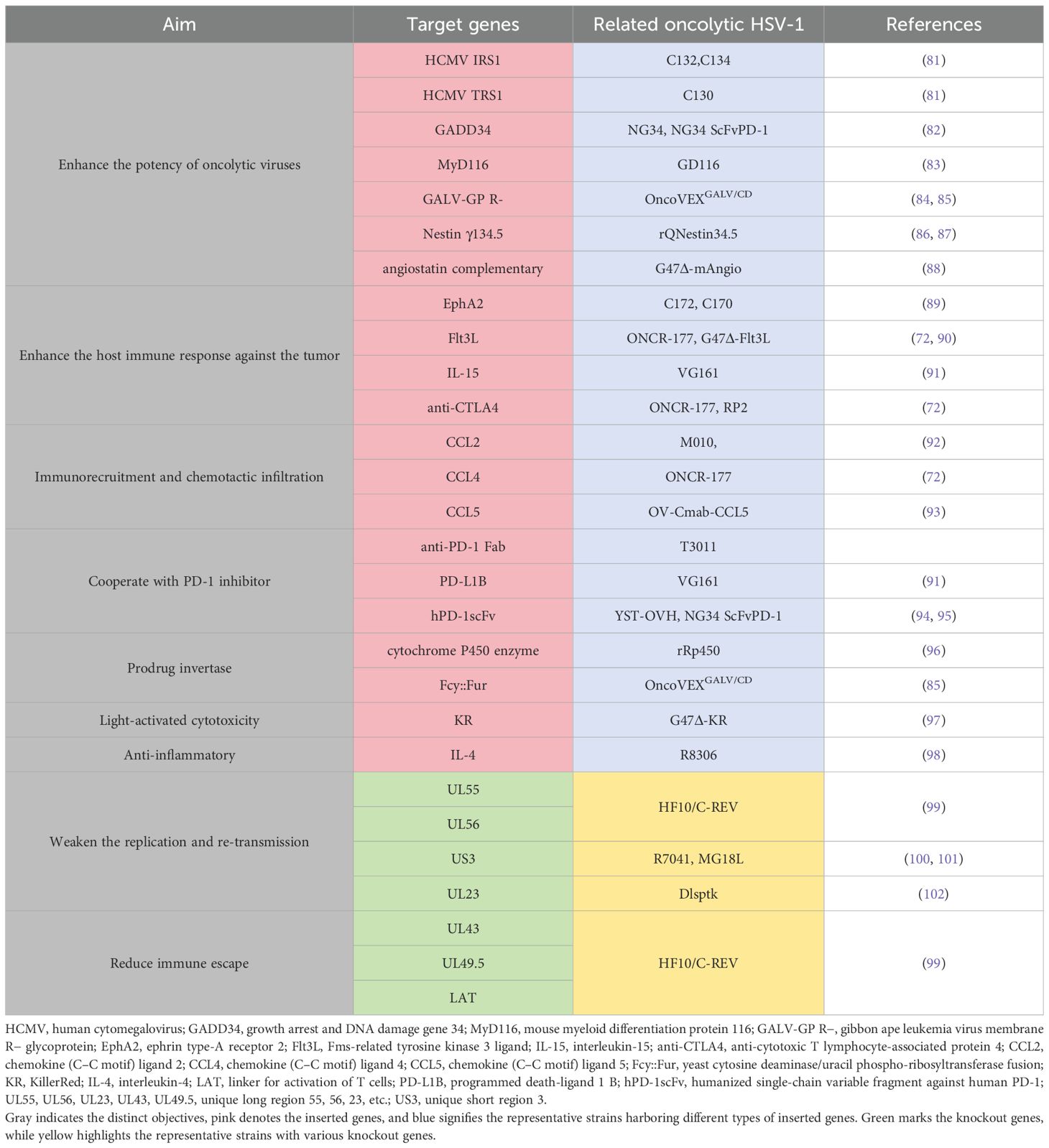
Table 1. Genetic modification of the modified oHSV.
GADD34 is homologous to γ134.5; like MyD116, it can substitute for γ134.5 to restore viral replication in glioblastoma and breast cancer cells, enhancing selective cytotoxicity (82, 83). The Gibbon leukemia virus fusion glycoprotein (GALV-GP) increases the efficiency of viral vector entry while inducing cell fusion, significantly boosting tumor cell death in vitro and promoting tumor shrinkage in vivo (84, 85). The Nestin promoter drives selective replication in glioma cells, enhancing glioma suppression when combined with cyclophosphamide (86, 87).
To induce and facilitate host immune responses against tumors, numerous attempts have been made to insert various genes into oncolytic viruses (OVs). EphA2 induces anti-tumor immunity by generating EphA2-specific CD8+ T cells that are effective against resistant tumors (89). Flt3L promotes dendritic cell development, thereby enhancing both local and systemic anti-tumor immune responses (90). IL-15 amplifies NK cell and CD8+ T-cell responses while enhancing tumor-specific immune cycles as demonstrated in pancreatic cancer models (91). Anti-CTLA4 antibody ONCR-177 increases the CD8+ T-cell response specific to tumor antigens, effectively inhibiting metastatic tumors while bolstering memory responses (103).
Some studies focus on immunorecruitment and chemotactic infiltration of immune cells into tumors to improve the efficacy of oncolytic viruses against malignancies. Chemokine genes such as CCL2, CCL4, and CCL5 are incorporated into OVs to enhance immune cell infiltration within tumors. For instance, a γ134.5-deficient HSV-1 expressing CCL2 along with IL-12 enhances glioma killing capabilities (92), whereas OV-CIMab-CCL5 improves outcomes in glioblastoma patients (93).
Certain genetic engineering studies target synergy with immune checkpoint inhibitors for enhanced anti-tumor effects. PD-1, associated synergistic genes inserted into the HSV genome, include single, stranded variable fragment PD-1 (ScFvPD-1), variable region components of antibodies targeting programmed death receptor one (anti-PD-1 Fab),and portions acting as PD -1 blockers (PD-L1B). Incorporating ScFvPD-1 sequences into NG34 virus augments anti-tumoral responses prolonging survival rates observed across ovarian carcinoma models alongside those exhibiting glioblastomas, demonstrating synergistic benefits when paired with PI3K inhibitors (94, 104). The ScFvPD-1 gene is also integrated within YST-OVH aiming at promoting systemic antitumoral reactions through CTLA–4 or TIM–3 blockade (95).
Another broad category concerning genetic modifications applied toward OV focuses upon prodrug activation mechanisms. Infected tumoral environments allow the synthesis of prodrug invertase produced intracellularly via virally encoded proteins, converting non-toxic precursors and directly transforming them into therapeutic agents. As early as 1998, cytochrome P450 was introduced within HSV-I, enabling conversion processes whereby cyclophosphamide becomes activated specifically inside malignant tissues, leading toward notable anticancer effects evidenced across medulloblastoma atypical teratoid/rhabdomyosarcoma brain neoplasms among others (105, 106). This approach yielded substantial advantages during treatment regimens involving diverse oncological conditions including but not limited to those previously mentioned (96, 107, 108).
An additional strategy involves inserting a gene-encoding yeast cytosine deaminase/uracil phospho-ribosyltransferase fusion(Fcy::Fur) into HSV-I, prompting infected neoplastic entities capable of synthesizing said construct. Fcy::Fur fusion catalyzes transformation processes wherein five-fluorocytosine (5-FC) is converted selectively, yielding toxic derivatives known as five-fluorouracil (5-FU), effectively targeting only malignant cellular populations without adversely affecting surrounding healthy tissue structures (85).
Recently, Kazuhide’s team successfully integrated killer red (KR) gene allowing light-induced singlet oxygen generation, which markedly enhanced overall effectiveness regarding treatments administered under laser irradiation particularly noted among cases involving both gliobastomatosis multiple myelomas (97).
To enhance safety profiles related specifically toward employing HSV-1-based therapeutics aimed at combating cancers, certain critical genomic deletions occur preventing uncontrolled propagation/infection events. Two primary methodologies exist focusing upon limiting risks tied closely together utilizing these engineered strains. One method entails restricting replicative capacity particle assembly through deletion, such as UL55, UL56, US3, and UL23, thus confining resultant virulence strictly localized around affected sites (99, 102, 109). Another tactic employs removing particular loci inclusive of UL43, UL49.5, and LAT, mitigating escape routes available and henceforth increasing the likelihood of successful elimination efforts directed toward residual pathogenic threats encountered post-treatment interventions (99).
5 Clinical trialsPreclinical studies have identified a substantial number of oHSVs with diverse antitumor properties. To gain a deeper understanding of the clinical application of oHSVs, we conducted a review of 34 published oHSV clinical trials spanning the past two decades (Table 2). Over half of these clinical trials were concentrated in Phases I and II, comprising 67% of the total. The three most common treatment methods were the injection of T-VEC, the combination of T-VEC injection and pembrolizumab, and the injection of G47Δ, which accounted for 26%, 11%, and 5.9% of the total, respectively. Among the tumors targeted by oHSV clinical trials, the top 2 were melanoma and brain tumors, representing 50% and 17.6% of the total, respectively. The oHSV type most frequently reported in clinical trials was T-VEC (n=22), accounting for 64% of all clinical trials. Notably, 22 out of the 34 clinical trials were conducted in the past 5 years, indicating a significant increase in research interest in this field.
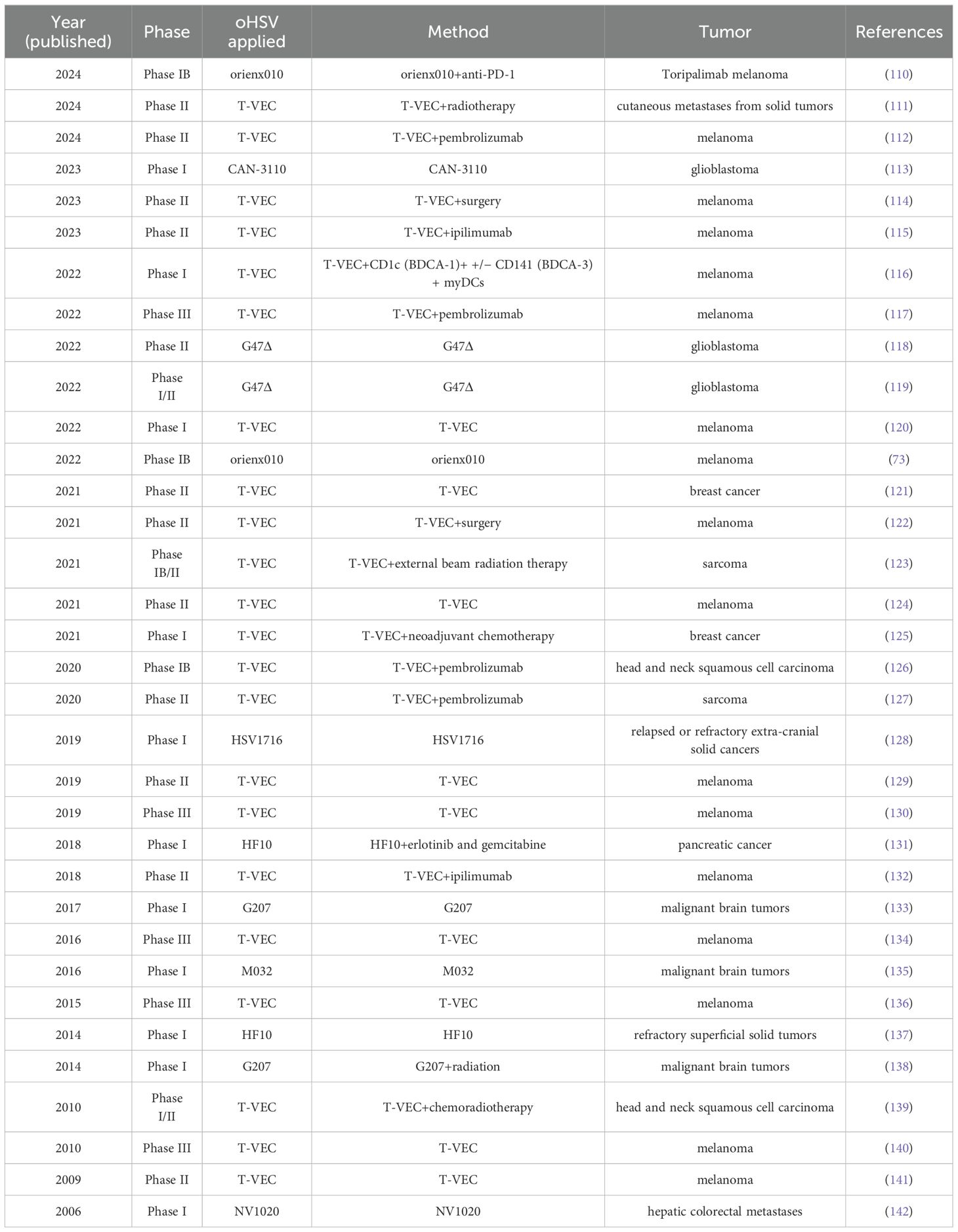
Table 2. Published clinical trials with oHSV.
6 DiscussionIn this review, we observed that the majority of oHSV clinical trials have employed various forms of viral modifications, such as deletions of genes γ134.5, US11, US12, and UL39, or the expression of transgenes like GM-CSF and IL-12. We also explored various gene modifications, which, despite not having been evaluated in clinical trials, represent a promising direction for future oncolytic virus research. Although oncolytic virotherapy is a promising anti-tumor technique, it is still facing several challenges.
The effectiveness of oncolytic viruses (OVs) is modest despite good safety. Viral genetic engineering improvements may enhance efficacy, but there are still obstacles in clinical trials, like balancing viral replication and immune responses, optimizing delivery routes, and achieving tumor-specific targeting.
During oncolytic virotherapy, it is imperative to achieve equilibrium between viral proliferation and the host’s anti-viral immune response. The ideal immune response is to allow viral replication early in oncolytic virotherapy and to initiate humoral immunity and clear the virus quickly at the end of treatment. The host immune system is crucial for tumor elimination but can clear OVs prematurely, limiting their therapeutic potential. Optimizing virus delivery and suppressing early immune responses give the virus more time for anti-tumor action. One of the strategies currently ongoing is to optimize delivery methods so that the virus moves silently into tumor cells before the host generates an immune response to clear the virus. Another strategy is to suppress the host immune response early on treatment, thereby improving the infection efficiency of the oncolytic virus. Upon completion of therapy, the introduction of antiviral medications expedites the virus’ elimination (143).
The current delivery methods include intratumoral injection and intravenous delivery. Intratumoral injection has the limitation of accessible tumors and is practically difficult in deep-seated or metastatic cases. For inaccessible tumors, imaging-guided or surgical approaches are required, which further complicate intratumoral injection. Intravenous delivery is more convenient than intratumoral injection. However, it requires high specificity to target tumors effectively, not to mention that it has risks of systemic toxicity and immune clearance.
Moreover, OVs as monotherapy may not achieve best therapeutic results. OVs are usually combined with other therapies, including immune checkpoint blockade or traditional anti-tumor therapies, to increase efficacy. Recently, integrating OVs with chimeric antigen receptor (CAR)-T cell therapy has emerged as an option. It could facilitate targeted delivery while improving bioavailability and enhancing tumor specificity. Furthermore, optimizing timing and dosing remains crucial for maximizing synergy between OVs and CAR-T cells (144, 145). A comprehensive regimen combining stereotactic body radiotherapy, oncolytic virotherapy, and pembrolizumab was used in clinical studies of metastatic non-small-cell lung cancer. The results demonstrate the superior prognosis of the comprehensive treatment regimen over conventional chemotherapy and pembrolizumab alone (146).
Potential safety issues of oncolytic virus therapy have also been suggested in clinical trials. For example, tumor cells died in large numbers after virus injection, resulting in the release of large amounts of antigenic material and cytokines. If the above-mentioned process occurs in a short time, it can lead to the life-threatening cytokine release syndrome. In addition, after the death of tumor cells, intracellular substances enter the circulation system and affect the coagulation system, which can lead to thrombosis or bleeding events. In addition, viruses may also cause insertional mutagenesis in host cells; for instance, the oncolytic adenovirus-based studies have found out the integration of viral genes into the host genome. As a kind of DNA virus, the possibility of insertional mutagenesis of HSV-1 virus is relatively small in theory, while long-term observation and studies are also needed toward this issue (147). Oncolytic HSV-1 has the potential to move through blood–brain barrier and infect the central nervous system, which, on the one hand, makes this type of oncolytic virus a candidate for the treatment of neurogenic malignancies, and on the other hand, increase the risk of central nervous system virus infection during the treatment of other tumors. Genetic modification is commonly used as one of the preventive strategies to reduce the pathogenicity of oncolytic viruses and improve their specificity for tumor cells. For example, G47Δ silenced γ134.5, UL39, US12, and US11 genes simultaneously (118). Clinical trials have shown that this kind of virus can barely replicate in vivo; therefore, treatment with the right dose of injected virus can safely treat tumors. Another preventive strategy is to combine oncolytic virus therapy with tumor immune checkpoint therapy or chemotherapy to kill the tumor while reducing the amount of oncolytic virus injection during the treatment. This strategy is currently widely used in clinical trial, such as the use of T-VEC virus strain combined with anti-PD-1 treatment (112, 126, 127).
As an increasing number of clinical trials explore newly engineered oncolytic virotherapies, these advancements are poised to yield significant breakthroughs in related research and promote the widespread adoption of oncolytic virotherapy for cancer treatment.
Author contributionsMZ: Writing – original draft, Writing – review & editing. ZS: Writing – original draft, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This article was supported by grants from the Natural Science Foundation of Hubei Province (No. 2022CFB286).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Santos AJ, Lima DSGV, Cordeiro SM, Silva LM, Silva SJ, Rocha PS, et al. Oncolytic virus therapy in cancer: A current review. World J Virol. (2021) 10:229–55. doi: 10.5501/wjv.v10.i5.229
PubMed Abstract | Crossref Full Text | Google Scholar
5. Agliardi G, Liuzzi AR, Hotblack A, De Feo D, Nunez N, Stowe CL, et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat Commun. (2021) 12:444. doi: 10.1038/s41467-020-20599-x
PubMed Abstract | Crossref Full Text | Google Scholar
8. Boldogkoi Z, Szucs A, Balazs Z, Sharon D, Snyder M, Tombacz D. Transcriptomic study of Herpes simplex virus type-1 using full-length sequencing techniques. Sci Data. (2018) 5:180266. doi: 10.1038/sdata.2018.266
PubMed Abstract | Crossref Full Text | Google Scholar
9. Goins WF, Wolfe D, Krisky DM, Bai Q, Burton EA, Fink DJ, et al. Delivery using herpes simplex virus: an overview. Methods Mol Biol. (2004) 246:257–99. doi: 10.1385/1-59259-650-9:257
PubMed Abstract | Crossref Full Text | Google Scholar
11. Ripa I, Andreu S, Lopez-Guerrero JA, Bello-Morales R. Interplay between autophagy and herpes simplex virus type 1: ICP34.5, one of the main actors. Int J Mol Sci. (2022) 23:13643. doi: 10.3390/ijms232113643
PubMed Abstract | Crossref Full Text | Google Scholar
12. Mao H, Rosenthal KS. Strain-dependent structural variants of herpes simplex virus type 1 ICP34.5 determine viral plaque size, efficiency of glycoprotein processing, and viral release and neuroinvasive disease potential. J Virol. (2003) 77:3409–17. doi: 10.1128/jvi.77.6.3409-3417.2003
PubMed Abstract | Crossref Full Text | Google Scholar
13. Cassady KA, Gross M, Roizman B. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J Virol. (1998) 72:8620–6. doi: 10.1128/JVI.72.11.8620-8626.1998
PubMed Abstract | Crossref Full Text | Google Scholar
14. Ahn K, Meyer TH, Uebel S, Sempe P, Djaballah H, Yang Y, et al. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. (1996) 15:3247–55. doi: 10.1002/j.1460-2075.1996.tb00689.x
PubMed Abstract | Crossref Full Text | Google Scholar
15. Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med. (1998) 187:341–8. doi: 10.1084/jem.187.3.341
PubMed Abstract | Crossref Full Text | Google Scholar
16. Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. (2017) 18:1–15. doi: 10.1007/s40257-016-0238-9
PubMed Abstract | Crossref Full Text | Google Scholar
17. Kanai R, Zaupa C, Sgubin D, Antoszczyk SJ, Martuza RL, Wakimoto H, et al. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol. (2012) 86:4420–31. doi: 10.1128/JVI.00017-12
PubMed Abstract | Crossref Full Text | Google Scholar
18. Pan C, Cai Q, Li X, Li L, Yang L, Chen Y, et al. Correction to: Enhancing the HSV-1-mediated antitumor immune response by suppressing Bach1. Cell Mol Immunol. (2022) 19:754. doi: 10.1038/s41423-022-00860-7
PubMed Abstract | Crossref Full Text | Google Scholar
19. Charron AJ, Ward SL, North BJ, Ceron S, Leib DA. The US11 gene of herpes simplex virus 1 promotes neuroinvasion and periocular replication following corneal infection. J Virol. (2019) 93:e02246–18. doi: 10.1128/JVI.02246-18
PubMed Abstract | Crossref Full Text | Google Scholar
20. Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells. vivo. Blood. (2011) 118:3890–900. doi: 10.1182/blood-2011-05-357111
PubMed Abstract | Crossref Full Text | Google Scholar
21. Kim K, Moon D, Kong SJ, Lee YS, Yoo Y, Kim S, et al. Antitumor effects of IL-12 and GM-CSF co-expressed in an engineered oncolytic HSV-1. Gene Ther. (2021) 28:186–98. doi: 10.1038/s41434-020-00205-x
PubMed Abstract | Crossref Full Text | Google Scholar
23. Lellahi SM, Azeem W, Hua Y, Gabriel B, Paulsen Rye K, Reikvam H, et al. GM-CSF, Flt3-L and IL-4 affect viability and function of conventional dendritic cell types 1 and 2. Front Immunol. (2023) 13:1058963. doi: 10.3389/fimmu.2022.1058963
PubMed Abstract | Crossref Full Text | Google Scholar
24. Minnar CM, Chariou PL, Horn LA, Hicks KC, Palena C, Schlom J, et al. Tumor-targeted interleukin-12 synergizes with entinostat to overcome PD-1/PD-L1 blockade-resistant tumors harboring MHC-I and APM deficiencies. J Immunother Cancer. (2022) 10:e004561. doi: 10.1136/jitc-2022-004561
PubMed Abstract | Crossref Full Text | Google Scholar
25. Bonfanti A, Lissoni P, Bucovec R, Rovelli F, Brivio F, Fumagalli L. Changes in circulating dendritic cells and IL-12 in relation to the angiogenic factor VEGF during IL-2 immunotherapy of metastatic renal cell cancer. Int J Biol Markers. (2000) 15:161–4. doi: 10.1177/172460080001500206
PubMed Abstract | Crossref Full Text | Google Scholar
26. Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. (1990) 250:1262–6. doi: 10.1126/science.2173860
PubMed Abstract | Crossref Full Text | Google Scholar
27. Zhang C, Tang J, Xie J, Zhang H, Li Y, Zhang J, et al. A conserved domain of herpes simplex virus ICP34.5 regulates protein phosphatase complex in mammalian cells. FEBS Lett. (2008) 582:171–6. doi: 10.1016/j.febslet.2007.11.082
PubMed Abstract | Crossref Full Text | Google Scholar
28. Cheng G, Gross M, Brett M, He B. AlaArg motif in the carboxyl terminus of the γ1 34.5 protein of herpes simplex virus type 1 is required for the formation of a high-molecular-weight complex that dephosphorylates eIF-2α. J Virol. (2001) 75:3666–74. doi: 10.1128/JVI.75.8.3666-3674.2001
PubMed Abstract | Crossref Full Text | Google Scholar
29. He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. P Natl Acad Sci USA. (1997) 94:843–8. doi: 10.1073/pnas.94.3.843
PubMed Abstract | Crossref Full Text | Google Scholar
30. He B, Gross M, Roizman B. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J Biol Chem. (1998) 273:20737–43. doi: 10.1074/jbc.273.33.20737
PubMed Abstract | Crossref Full Text | Google Scholar
31. Li Y, Zhang C, Chen X, Yu J, Wang Y, Yang Y, et al. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J Biol Chem. (2011) 286:24785–92. doi: 10.1074/jbc.M111.232439
留言 (0)