
記住我
Messenger RNA (mRNA) vaccines have emerged as a rapid, flexible, and scalable strategy in cancer immunology. This innovative method elicits a robust and targeted immune response (Lorentzen et al., 2022; Yao et al., 2024). The effectiveness of mRNA vaccines during the COVID-19 pandemic has underscored their potential in addressing infectious diseases (Chakraborty et al., 2021; Pennisi et al., 2024). However, moving from concept to clinical implementation involves navigating significant scientific and technical challenges, necessitating a comprehensive, interdisciplinary approach (Lorentzen et al., 2022; Sayour et al., 2024). mRNA vaccines in oncology are considered personalized, representing a key advance in precision medicine by targeting the unique genetic mutations in an individual’s tumor cells (Lorentzen et al., 2022). By crafting a vaccine that targets these specific anomalies, this personalized method seeks to elicit a precise immune response, minimizing off-target effects and significantly enhancing therapeutic outcomes (May, 2024).
Unlike traditional vaccines, which use inactivated or attenuated pathogenic proteins, mRNA vaccines deliver tumor-associated antigens (TAAs) or neoantigens directly to antigen-presenting cells (APCs) like dendritic cells (DCs) or macrophages. After the tumor antigen is presented on the surface of the APCs, a cascade of immune responses is triggered, initiating adaptive immunity (Pardi et al., 2018; Gote et al., 2023). These neoantigens are processed and displayed on the cell surface via major histocompatibility complex (MHC) class I molecules, allowing the immune system to recognize the tumor proteins as foreign and triggering an immune response (Esprit et al., 2020). The primary immune response involves cytotoxic T lymphocytes (CTLs), which recognize and eliminate cancer cells expressing specific tumor antigens (Vishweshwaraiah and Dokholyan, 2022). Additionally, APC activation stimulates CD4+ T helper 1 (TH1) cells, which release cytokines to boost CTL activity and recruit macrophages, creating an immune-reactive tumor microenvironment (TME) (Li et al., 2022b; Ramirez et al., 2023). By enhancing the infiltration of immune cells, such as CTLs and macrophages, and overcoming immune checkpoint inhibition, mRNA vaccines can help shift the balance in favor of anti-tumor immunity. This reprogramming of the TME supports a more effective and sustained immune response against cancer cells, ultimately improving the overall efficacy of cancer immunotherapy (Gote et al., 2023; Ramirez et al., 2023).
In the case of naked mRNA vaccines, the mRNA is delivered directly into the body without any protective carrier. Once administered, the naked mRNA is taken up by cells, including DCs, through endocytosis or direct membrane fusion (Hasan et al., 2023). After entering the cytoplasm, the mRNA is translated into the target tumor antigen, which is processed and presented on MHC class I molecules, stimulating a robust immune response, specifically activating CTLs that target and destroy tumor cells expressing the same antigen. However, naked mRNA has some limitations, particularly in terms of stability and delivery efficiency (Abbasi et al., 2024).
To overcome these challenges, lipid nanoparticles (LNPs)-encapsulated mRNA are commonly used, and they are the only FDA-approved delivery vehicles for mRNA vaccines (Igyártó and Qin, 2024). LNPs are designed to encapsulate the mRNA, protecting it from degradation and improving its stability in the bloodstream. They also facilitate the efficient delivery of mRNA into target cells. Once inside the cell, the mRNA is released from the LNPs and enters the cytoplasm, where translation occurs, leading to the production of tumor antigens. LNPs are especially advantageous for improving cellular uptake. They interact with the cell membrane, facilitating endocytosis and ensuring that the mRNA is delivered into cells in a controlled manner. Once inside, the mRNA is translated into the antigen, processed, and presented by APCs on MHC class I molecules, leading to the activation of CTLs and the initiation of a strong anti-tumor immune response (Imani et al., 2024). By using LNPs, the delivery of mRNA vaccines becomes more efficient, enhancing both the stability of the mRNA and the ability of APCs to initiate a targeted immune response (Alameh et al., 2021; Shuptrine et al., 2024).
The development of personalized mRNA vaccines involves several crucial steps, each supported by advanced bioinformatics tools. Initially, next-generation sequencing (NGS) is used to analyze the genome of the pathogen or tumor, identifying unique mutations and neoantigens (Alburquerque-González et al., 2022; Al Fayez et al., 2023). Comprehensive genetic data is crucial for designing mRNA vaccines. Tools like NetMHCpan and the Immune Epitope Database (IEDB) identify the most immunogenic HLA-I and MHC class I epitopes to trigger a strong T-cell response (Kim et al., 2012; Cai et al., 2021). To enhance stability and efficiency, RNAfold and mfold predict the mRNA’s secondary structure, reducing degradation and improving effectiveness (Chen and Chan, 2023). LNP formulation tools, such as NanoAssembler, optimize delivery by protecting the mRNA and aiding its entry into host cells for effective antigen expression (Wang et al., 2022).
On the other hand, machine learning algorithms further refine these predictions by analyzing extensive immunological data. Incorporating machine learning and AI into this process is vital. Algorithms like Random Forest, Support Vector Machines (SVMs), and Convolutional Neural Networks (CNNs) analyze large datasets to predict vaccine efficacy and potential side effects. These AI-driven insights help optimize vaccine design, enhancing efficacy and safety (Bravi, 2024).
While advancements in bioinformatics and AI are significant, comprehensive comparative studies in this field are lacking, which limits our understanding of their full potential. This paper explores the role of these technologies in developing personalized mRNA vaccines, focusing on genome sequencing, epitope prediction, RNA structure analysis, and LNP formulation. We discuss the challenges, insights, and future directions, highlighting how AI improves vaccine development by analyzing data, identifying patterns, and optimizing design to predict side effects and enhance effectiveness. This paper aims to address current knowledge gaps and encourages further research in oncology and immunology, where personalized mRNA vaccines have the potential to transform cancer treatment.
2 Sequencing and initial data acquisitionFigure 1 presents a schematic overview of bioinformatics tools for mRNA structure prediction and design, covering methods for secondary structure prediction, coding sequences (CDS) optimization, and 3D modeling. Sequencing and initial data acquisition are fundamental steps in developing mRNA vaccines, providing essential genetic information about target viruses, and setting the stage for vaccine design and optimization (Gunter et al., 2023). Key sequencing technologies such as Illumina, Oxford Nanopore, and PacBio play crucial roles in this process. Illumina’s high-throughput short-read sequencing offers extensive coverage of viral genomes, helping to identify genetic variations that are important for understanding viral diversity and evolution (Lemay et al., 2022). Oxford Nanopore’s real-time long-read sequencing provides insights into full-length RNA transcripts and complex genomic regions, which is useful for detecting diverse viral variants and structural features (Stefan et al., 2022). PacBio’s high-accuracy long-read sequencing allows for detailed genomic characterization and variant analysis, particularly beneficial for studying RNA viruses. Bioinformatics tools play a crucial role in maintaining the quality, preprocessing, and comprehensive analysis of sequencing data across all three sequencing technologies. FASTQC (Fast Quality Control) assesses key quality metrics like base quality scores and GC content, while Trimmomatic eliminates artifacts and adapter sequences from raw reads (Bolger et al., 2014), thereby improving the accuracy of subsequent analyses. SAMtools manages aligned sequences in Sequence Alignment/Map (SAM) and Binary Alignment/Map (BAM) formats, which is vital for variant calling and in-depth genomic analysis, offering valuable insights for vaccine design. The workflow begins with alignment tools such as the Burrows-Wheeler Aligner (BWA) and Bowtie, which align short-read mRNA sequences to reference genomes or transcriptomes (Rajan-Babu et al., 2021). Also, Visium Spatial Gene Expression (Visium SGE) is an advanced platform that combines spatially resolved transcriptomics with histological imaging to map gene expression within the structural context of tissues, enabling precise insights into cellular activity and tissue architecture (Toyama et al., 2023). These tools are instrumental in identifying conserved regions and potential immunogenic epitopes within the mRNA sequences. Following alignment, assembly algorithms reconstruct full-length mRNA sequences by integrating sequence overlaps and pairing information, ensuring the integrity and completeness of mRNA constructs for vaccine production.
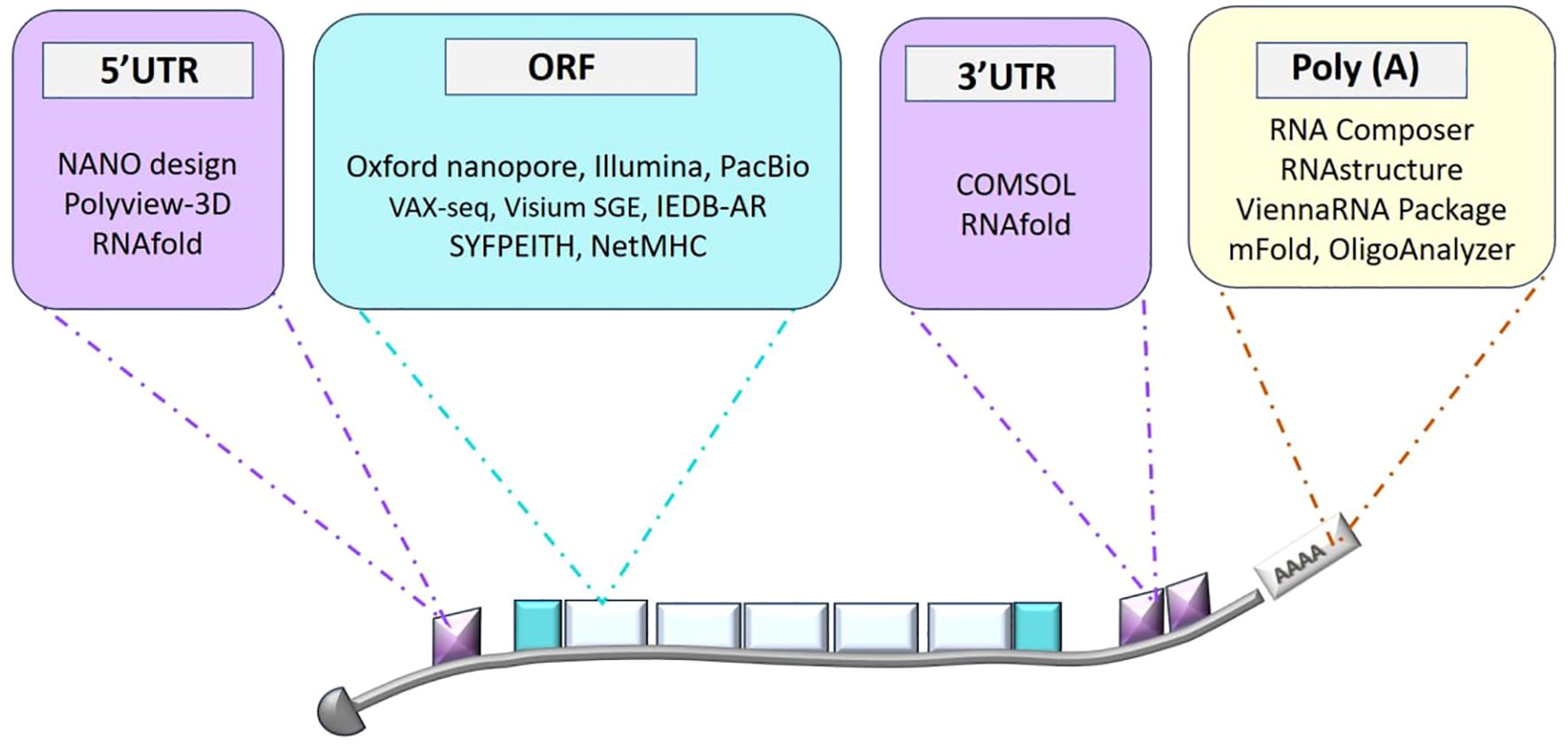
Figure 1. Overview of bioinformatics tools for mRNA structure prediction and design. This diagram highlights the various stages of mRNA design, including secondary structure prediction, coding sequence (CDS) optimization, and 3D structure modeling, along with the bioinformatics tools employed at each stage to enhance mRNA design for therapeutic applications.
In practice, during the development of mRNA vaccines for novel viral outbreaks, alignment tools like BWA are used to compare mRNA sequences with known sequences of related viruses. This process helps identify conserved regions critical for vaccine design, ensuring effective targeting of the virus and the induction of protective immune responses in vaccinated individuals. Table 1 compares the main key sequencing technologies, including Illumina, Oxford Nanopore, and PacBio.
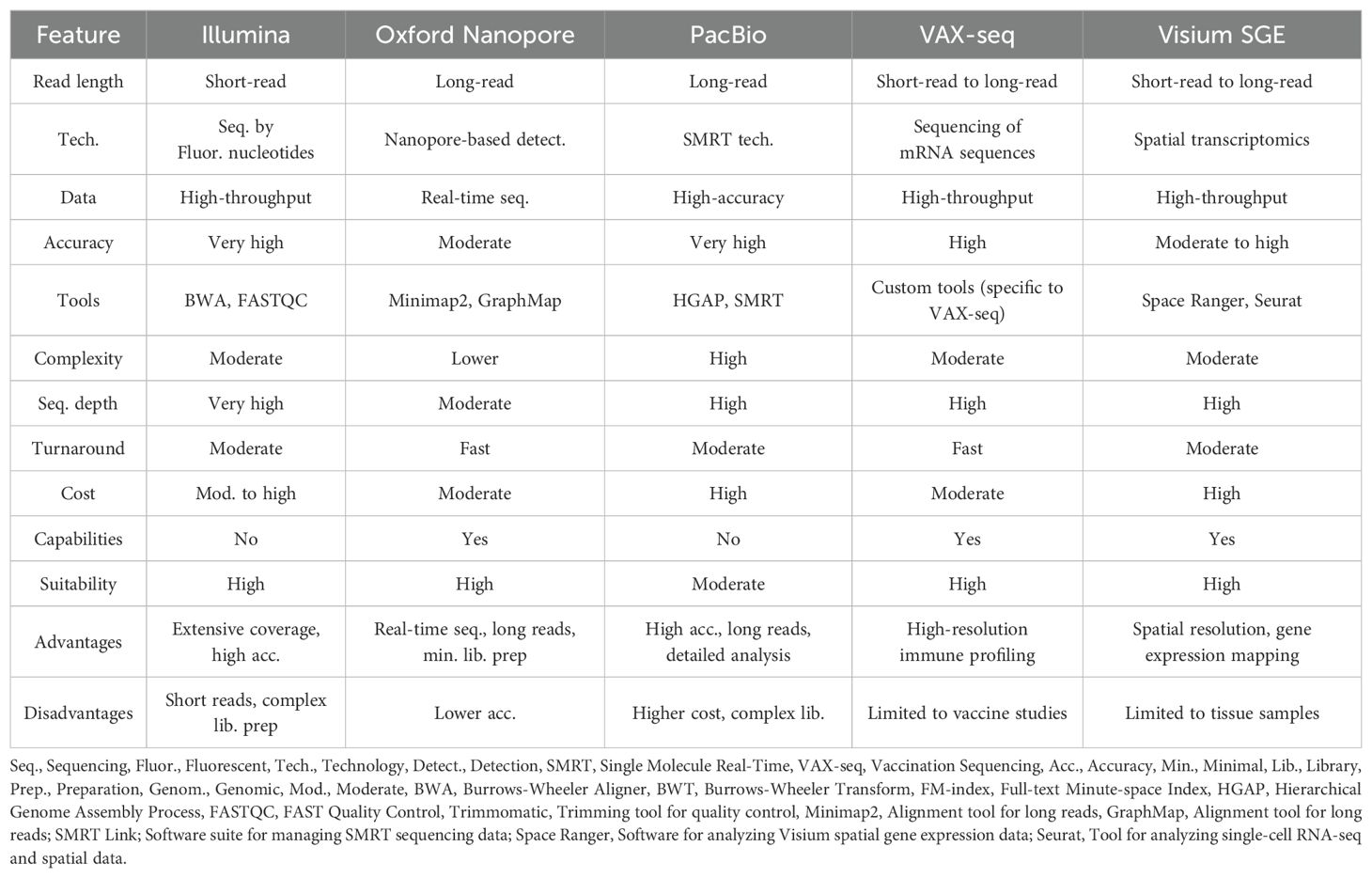
Table 1. Comparison of sequencing technologies for mRNA vaccine development.
2.1 IlluminaIllumina sequencing is a high-throughput technology known for its precision in generating short DNA or RNA sequence reads, which can produce fragments hundreds of bases in length and is vital for mRNA vaccine development. The process starts with fragmenting DNA samples of approximately 300-500 bp or RNA samples of about 200 bp, followed by the attachment of adapters. These fragments are then amplified on a flow cell through bridge amplification, forming clusters of identical sequences. During sequencing, fluorescently labeled nucleotides are incorporated into the growing DNA strands. Techniques like RELIC are used to correct dye bias in Illumina data, ensuring accurate sequencing results (Xu et al., 2017). Illumina can also help length-sequencing platforms such as ONT get high-quality genomes more efficiently (Lerminiaux et al., 2024). Each nucleotide emits a unique color when excited by a laser, and high-resolution cameras capture these colors to determine the nucleotide sequence. This technology is crucial for identifying genetic variations and viral genome features, aiding in the development of effective mRNA vaccines. For processing sequencing data, algorithms like BWA (Guo and Huo, 2024) and Bowtie are essential. BWA uses the Burrows-Wheeler Transform (BWT) (Keel and Snelling, 2018) for efficient sequence alignment, while Bowtie utilizes the FM-index for indexing and searching data (Table 1). Brittney N. Keel’s comparison shows that BWA is more robust, whereas HISAT2 is faster and uses less memory than both BWA and Bowtie2 (Keel and Snelling, 2018). Data quality is ensured with preprocessing tools such as FASTQC, which evaluates base quality scores, and Trimmomatic, which removes low-quality bases and adapter sequences to enhance alignment accuracy and variant detection. Detailed mathematical formulations and specific operational details of these methods are available in Supplementary file S1.
2.2 Oxford nanoporeOxford Nanopore sequencing is a state-of-the-art long-read technology that plays a crucial role in mRNA vaccine development. This method uses nanopore sensors to detect changes in ionic current as nucleic acids pass through a protein nanopore (Xue et al., 2020), such that a negatively charged single-stranded DNA or RNA molecule is driven from the negatively charged “cis” side through the nanopore to the positively charged “trans” side, which is recorded and analyzed to infer the base sequence (Su et al., 2023). Currently, there are eight versions of the system, with R9 achieving an impressive translocation rate of 250 bases per second and R9.4 achieving a translocation rate of 450 bases per second, which is a significant improvement over R7’s 70 bases per second. The other different systems have their advantages (Wang et al., 2021).
For mRNA vaccine development, Oxford Nanopore sequencing has been modified to sequence them directly without reverse transcription. Although the accuracy of direct sequencing of RNA is lower than that of DNA sequencing, about 83% to 86%. Similarly, Oxford Nanopore sequencing also provides direct sequencing of complementary DNA (cDNA) without the need for polymerase chain reaction (PCR) amplification (Wang et al., 2021). This capability is essential for understanding the complete structure and function of RNA, including secondary structures and complex genomic regions vital for designing effective vaccines.
Key bioinformatics tools for Oxford Nanopore sequencing are MinION Knowledge Base (MinKNOW) and Guppy. MinKNOW manages the sequencing device and collects raw data (Oeck et al., 2023), while Guppy performs base-calling to convert the raw signal data into nucleotide sequences (Wick et al., 2019). After base-calling, alignment tools such as Minimap2 are used to map these long reads to reference genomes. During mRNA vaccine development, this technology allows real-time sequencing of viral genomes, aiding in the identification of conserved regions and potential epitopes crucial for effective vaccine design. Although the average accuracy of ONT sequencing is improving, certain subsets of reads or read fragments have very low accuracy, and the error-rate reads of 1D reads and 2D/1D reads are still much higher than the short reads produced by NGS technologies (Wang et al., 2021). Oxford Nanopore sequencing also excels at detecting viral variants by analyzing complete sequences and complex genomic regions, with the characteristics of short turnaround time and low cost (Xu et al., 2022). This ability to identify mutations and variations is essential for designing mRNA vaccines that elicit strong immune responses against diverse viral strains.
2.3 PacBioPacBio sequencing, utilizing Single Molecule Real-Time (SMRT) targeting technology that does not require pausing between read steps, so kinetic changes interpreted from light-pulse movies can be analyzed to detect base modifications, such as methylation, and accurate detection and discovery of all variant types, even in hard-to-reach regions of the genome (Rhoads and Au, 2015), has the potential to revolutionize physical health, reproduction, cancer research, as well as microbial and viral genetic testing (Ardui et al., 2018), is crucial for mRNA vaccine development due to its capability to produce highly accurate long-read sequences. This technique involves DNA polymerase synthesizing complementary DNA strands with fluorescently labeled nucleotides. The emitted light from these nucleotides is detected in real-time, enabling immediate base calling. For RNA molecules, PacBio finds novel genes, transcripts, and alternative splicing through a complete view of transcript isoform diversity to sequence them (Rhoads and Au, 2015).
The long-read capability of PacBio sequencing, which can extend up to 60 kb, provides significant advantages in identifying and quantifying subtypes, including novel ones (Rhoads and Au, 2015). According to Jia H. et al. findings, this technology allows for low-input library preparation, requiring only 100 ng of DNA for the Sequel system and 400 ng for the Sequel II system (Jia et al., 2024). This is particularly useful for comprehensive viral genome sequencing, including the identification of new variations and genetic mutations in viruses like SARS-CoV-2 (Nicot et al., 2023).
The SMRT Link software suite manages data collection and processing, including base calling and error correction. Algorithms such as the Hierarchical Genome Assembly Process (HGAP) (Chin et al., 2013) and Canu et al. (Prjibelski et al., 2023) address the challenges of assembling long reads by correcting errors and constructing complete genome sequences. HGAP builds consensus sequences from long reads. PacBio sequencing is crucial for identifying conserved regions and potential immunogenic epitopes within viral genomes, which helps in designing effective mRNA vaccines. However, the technology has limitations, including lower throughput with fewer sub-reads or CCS reads and a higher error rate of about 11-15% for CLR reads (Rhoads and Au, 2015).
2.4 VAX-seqVAX-seq, a novel sequencing technology, plays a pivotal role in advancing the field of mRNA vaccine development. This high-throughput sequencing method is specifically tailored for the identification and quantification of vaccine-induced immune responses (Gunter et al., 2023b). VAX-seq is a specialized technology focused on sequencing mRNA in the context of immune profiling. Its ability to detect modified nucleosides is limited and primarily inferred through indirect analyses or complementary assays (Gunter et al., 2023). By providing a more detailed understanding of the interactions between mRNA vaccines and the immune system, VAX-seq enables the identification of specific mRNA sequences that contribute to optimal immune activation. This technology allows researchers to profile the genetic composition of mRNA vaccines and their translation products with greater accuracy, improving both the design and efficacy of these vaccines (Gote et al., 2023).
One of the key advantages of VAX-seq over traditional sequencing methods, such as Illumina and Oxford Nanopore, lies in its ability to offer higher-resolution insights into the transcriptome (Gunter et al., 2023). This enables a more comprehensive analysis of vaccine-induced responses, allowing for the detection of rare or subtle immune reactions that might be missed with other methods. The technique enhances the ability to tailor mRNA vaccine sequences to better stimulate desired immune responses, which is crucial for optimizing vaccine formulations for various pathogens, including those that require more precise immune targeting. Incorporating VAX-seq into mRNA vaccine development holds significant potential for both enhancing vaccine design and guiding clinical decision-making. By combining its high sensitivity with the ability to sequence and quantify complex mRNA sequences, VAX-seq aids in the identification of critical sequence motifs and epitopes (Jeeva et al., 2021). This level of detail is essential for the development of more effective mRNA vaccines, capable of eliciting stronger, more targeted immune response, and ultimately providing better protection against infectious diseases (Gunter et al., 2023).
2.5 Visium SGEVisium SGE by 10x Genomics has emerged as a transformative analytical tool, integrating spatially resolved transcriptomic data with high-resolution tissue histology (Ståhl et al., 2016). This platform allows researchers to map gene expression patterns directly onto histological sections, providing unparalleled insights into the spatial context of mRNA translation and immune cell dynamics within tissues (Toyama et al., 2023). Visium SGE combines spatial transcriptomics with high-throughput short-read sequencing. While it offers spatial resolution and gene expression mapping, its capability to detect modified nucleosides is restricted to indirect bioinformatic inferences (Williams et al., 2022). By combining transcriptomics with histopathological features, Visium enables the identification of specific cell populations and their molecular activities about their precise tissue location. For example, in the context of mRNA vaccine development, Visium can localize mRNA-encoded antigen expression to immune-competent regions, such as lymphoid aggregates, while simultaneously identifying structural changes in surrounding tissue architecture. This dual-layer information is invaluable for validating predictive models like AlphaFold, ensuring that computationally predicted antigens are accurately expressed and situated in biologically relevant microenvironments (Smith et al., 2024). In mRNA vaccine development, Visium has proven instrumental in refining antigen design and delivery strategies. For instance, studies leveraging Visium have demonstrated its capability to map DCs activity in lymphoid tissues following mRNA-LNP administration, directly linking antigen presentation to CTLs recruitment. In one example, Visium analysis identified specific tissue regions where mRNA vaccines encoding TAAs were translated most efficiently, allowing researchers to pinpoint the spatial co-localization of antigen-expressing cells and CD8+ T-cell activation zones. This spatial information guided the optimization of LNP formulations to ensure antigen delivery to DCs located in lymphoid-rich areas, thereby enhancing CTL priming and overall vaccine efficacy (Melo Ferreira et al., 2021; Hudson and Sudmeier, 2022).
Moreover, Visium facilitates the identification of off-target effects and unintended mRNA expression in non-target tissues, a critical consideration in vaccine safety profiling. For example, spatial transcriptomic analysis using Visium uncovered ectopic expression of mRNA constructs in hepatocytes during preclinical studies, revealing suboptimal LNP biodistribution. Based on these findings, LNP formulations were redesigned to incorporate specific targeting ligands that preferentially deliver mRNA to DCs while minimizing liver uptake (Ståhl et al., 2016). This iterative approach underscores the power of Visium in bridging computational predictions with experimental outcomes, ensuring the spatial fidelity of mRNA expression, and advancing the rational design of mRNA vaccines for cancer immunotherapy (Toyama et al., 2023).
3 Antigen and epitope predictionAntigen prediction uses bioinformatics to analyze pathogen genomes or proteomes, identifying specific epitopes that trigger immune responses through various MHC classes or DC and macrophages (Capelli et al., 2023). In mRNA vaccine development, choosing the right antigen targets is essential for effective expression and a strong immune response. Neo-antigen prediction technologies enhance vaccine safety and effectiveness by finding highly immunogenic epitopes, which can shorten development timelines and reduce costs (Soria-Guerra et al., 2015). The following section will detail the specific tools used in this process. Selecting optimal epitopes is crucial for robust immune stimulation, and epitope prediction tools are key in developing effective mRNA vaccines.
3.1 NetMHCNetMHC is a user-friendly bioinformatics tool that utilizes information from both data types for training on binding affinity and eluting ligand data, thus being used to predict peptide-MHC interactions, addressing the challenge of identifying peptides that effectively bind to MHC molecules. NetMHC has undergone several transformative updates since its inception in the early 2000s, embracing the latest computational advancements and significantly enhancing its database of interactions between peptides and MHC. These updates have incorporated sophisticated scoring matrices, intricate hidden Markov models, and cutting-edge artificial neural networks (ANNs) (Zhou et al., 2023), collectively enhancing the tool’s predictive capabilities and broadening its application scope within the field, for example, NetMHCpan-4.0 achieves better performance, and ligands in all cases are predicted with very strong eluting ligand likelihood values (Jurtz et al., 2017). It has become an essential resource in immunoinformatics, crucial for understanding how peptide fragments derived from pathogens can activate CD8+ T cells and trigger immune responses, particularly neoantigens in cancer immunology (Wu et al., 2023).
Cytotoxic T cells play a central role in the pathogenesis and immunomodulation of malignancies, and the binding of peptides to MHC molecules is the most selective single step in the antigen presentation pathway. It has recently been shown that over 90% of naturally occurring MHC ligands are identified with 98% specificity (Nielsen and Andreatta, 2016). In vaccine development, NetMHC evaluates the binding affinity between peptides and MHC molecules, aiding researchers in selecting optimal peptides for vaccine inclusion to induce robust CD8+ T-cell responses. This capability enhances vaccine specificity and efficacy by focusing on peptides with the strongest interactions. During the intricate process of vaccine development, the versatile NetMHC tool harmoniously integrates with existing peptide-MHC data, leveraging computational simulations to accurately predict potential antigen epitopes – a pivotal step in vaccine design. This training approach integrates larger data content and can directly learn the length of each MHC molecule from the experimental binding data to present the optimal peptide (Andreatta and Nielsen, 2016). NetMHC provides highly accurate predictions due to its use of extensive training datasets and advanced modeling techniques like neural networks and position-specific scoring matrices (PSSMs). While it focuses on MHC class I molecules, its performance depends on the quality and breadth of peptide-MHC interaction data and may require substantial computational resources. Although NetMHC is excellent at predicting MHC interactions, it does not cover all aspects of antigen processing and presentation, such as class II MHC interactions. Despite these limitations, integrating NetMHC into the vaccine development process greatly improves the design of specific and effective vaccines.
3.2 IEDB Analysis ResourceThe IEDB-AR (Immune Epitope Database Analysis Resource) is a crucial tool for designing mRNA vaccines against variable antigens, especially for virus-based vaccines such as those targeting influenza viruses, SARS-CoV-2, and HIV.I EDB-AR has T cell epitope prediction tools, B cell epitope prediction tools, and tools for the analysis of known epitope sequences or sequence groups. The IEDB-AR platform stands as an ideal choice for addressing diseases characterized by substantial antigenic variation or requiring a robust, multifaceted immune response. Its applicability extends to a wide spectrum of conditions, including those associated with infections, allergies, autoimmune disorders, and transplantations, where its capabilities are particularly well-suited to inform and guide therapeutic strategies (Vita et al., 2018).
Several new tools have been added to IEDB-AR. Among the T cell epitope prediction tools are TepiTool, MHC-NP, Immunogenicity, CD4EpiScore, and Deimmunization. These tools have their different functions, such as TepiTool, It can be used to predict naturally processed MHC class I and II ligands, deimmunization of therapeutic proteins, and prediction of T cell immunogenicity beyond MHC binding affinity (Dhanda et al., 2019). IEDB-AR also adds a new tool called LYRA (Automated Modeling of Lymphocyte Receptors), which allows for the simulation of 3D structures of B and T cell receptors (Klausen et al., 2015), allowing for the prediction of canonical structures per cycle, when necessary.
By utilizing algorithms such as ANNs and SVMs to predict both class I and II peptide-MHC binding affinities, T-cell and B-cell epitopes, and cross-reactive epitopes (Yan et al., 2024), IEDB-AR identifies optimal antigenic targets, such as pHLA-target Ags (Gerber et al., 2020), to stimulate both CD4+ and CD8+ T-cell responses, as well as antibody responses. However, its effectiveness depends on the quality and comprehensiveness of the peptide-MHC interaction data, which can impact prediction accuracy. This variability in data coverage may affect the tool’s precision.
3.3 SYFPEITHISYFPEITHI, a free bioinformatics tool from the late 1990s, predicts peptide-MHC interactions for MHC class I and II molecules. Its user-friendly interface and high accuracy help identify peptides that bind to specific MHC molecules and predict epitopes (Zhang et al., 2023a). The database includes peptide sequences (approximately 200 peptide motifs and 2000 peptide sequences), anchor position, MHC specificity, source protein, source organism, and publication references. The tool employs PSSMs as its primary algorithm to evaluate the binding affinity of peptides to MHC molecules, which can sequence the MHC-eluting peptides directly. The adopted scoring approach simplifies the identification of promising vaccine candidates by providing detailed binding scores and rankings. Chao Shen et al. findings show that this method effectively balances scoring and docking tasks, making the selection process both rigorous and efficient (Shen et al., 2023). But instead of synthesizing and testing dozens or even hundreds of peptides, SYFPEITHi prescreens a set of peptides and enables epitope prediction of the sequence, restriction elements, and their respective motifs of proteins or their genes., which aids in the design of effective vaccines (Rammensee et al., 1999). The accuracy of SYFPEITHI’s predictions depends on the quality and completeness of the peptide-MHC interaction data, with gaps potentially affecting reliability. SYFPEITHI does not account for critical aspects of antigen processing and presentation, such as peptide transport into the endoplasmic reticulum via TAP, proteasome trimming, or competition for MHC binding. These factors are essential for a full understanding of immune responses and peptide presentation (Larsen et al., 2005; Lee et al., 2024a).
4 Codon optimizationBefore analyzing mRNA structure, it’s essential to focus on the Coding Sequence (CDS) and codon optimization. Codon optimization is crucial for improving CDS expression in a host organism. This process involves modifying codons to match the host’s preferred codon profile, which enhances gene expression efficiency and reduces costs (Hanson and Coller, 2018). Codon optimization takes into account factors such as codon usage bias, tRNA abundance, GC content, and RNA secondary structure. By carefully selecting codon combinations, researchers can improve protein expression, reduce mRNA degradation, and enhance stability. This also impacts protein folding, post-translational modifications, and immunogenicity (Zhang et al., 2023b). Software tools like GeneOptimizer and JCAT (Java Codon Adaptation Tool) help in this process by choosing the most efficient codons based on the host’s tRNA abundance and codon usage patterns. Here is a summary of their advantages and disadvantages.
4.1 GeneOptimizerGeneOptimizer is a powerful tool for optimizing DNA sequences. It uses a sliding window method to adjust codon usage, GC content, and other factors to improve translation efficiency (Fu et al., 2020). It handles large gene sequences and manages key processes such as transcription, splicing, translation, and mRNA degradation. GeneOptimizer can complete gene optimization in minutes. Synthetic genes were designed by uploading sequences, selecting expression systems, specifying cloning vectors, and sequence details. At the same time, based on the data related to a given organism and the user’s sequence requirements, the DNA sequence that is most suitable for the user’s research requirements is generated. Researchers can use this tool to select optimal codon combinations for specific organisms, enhancing gene expression efficiency and scaling up protein production to meet experimental needs. Despite being a premium tool, GeneOptimizer empowers users with the autonomy to meticulously craft gene sequences, circumventing the necessity for DNA templates. It achieves this through the implementation of sophisticated codon optimization and sequence alignment algorithms, exemplified by its utilization of sliding windows for refining multiparameter DNA sequences and FOGSAA for executing swift, global sequence alignments (Chakraborty and Bandyopadhyay, 2013). Importantly, GeneOptimizer enhances mRNA stability and prolongs its half-life within cells through codon optimization (Schwanhäusser et al., 2011; Luo et al., 2023). Optimized mRNA sequences, with more favorable codons, related studies have shown that using GeneOptimizer at the same dose can significantly increase protein expression and produce more antigen proteins, leading to stronger immune responses and improved disease prevention. However, altering mRNA sequences with GeneOptimizer may have some unknown risks, such as potential interactions with other RNA and proteins within cells, which could lead to adverse reactions or reduced vaccine efficacy.
4.2 JCATThe JCAT uses advanced algorithms, such as the Codon Adaptation Index and the Relative Codon Adaptation model, to enhance the production of heterologous proteins and there is no need to manually define highly expressed genes. Significantly, JCAT not only enhances gene sequence design but also safeguards against undesirable outcomes such as the emergence of restriction enzyme cleavage sites and Rho-independent transcription terminators. Grote et al.’s study underscores this capability, demonstrating how JCAT successfully adapted the codon usage of the P. aeruginosa exbD gene to that of E. coli while simultaneously evading the formation of identical restrictive sites, ensuring the stability of CDS. On the output, JCAT can be either a graph or a CAI (Codon Adaptation Index) value given by the pasted sequence and the newly adapted sequence. In addition, users can calculate CAI values by uploading gene sequences in FASTA format (Grote et al., 2005), which can help researchers quickly understand key biological information during mRNA vaccine development. JCAT is usually a codon optimization of a single gene in the laboratory. Therefore, experiments are comparing the original Pseudomonas aeruginosa DNA sequence with the DNA sequence optimized for Escherichia coli to demonstrate the degree of optimization. JCAT is user-friendly, offering high automation and precision, which allows researchers to efficiently analyze and adjust codon combinations. This optimization improves mRNA vaccine expression levels in host cells and avoids Rho-independent transcription terminators in codon-optimized DNA sequences (Postle and Good, 1985; Ermolaeva et al., 2000).
JCAT is built on biological insights into translational optimization, particularly the significance of codon adaptation in heterologous protein production. By leveraging algorithms like the Codon Adaptation Index (CAI), it aligns codon usage with host-specific tRNA pools, improving translation efficiency and reducing translational errors (Sample et al., 2019). Studies have shown that codon optimization not only enhances protein yield but also stabilizes mRNA expression by avoiding undesired sequence features, such as Rho-independent transcription terminators, which can destabilize transcripts (Leppek et al., 2022). Biologically, JCAT addresses critical factors in mRNA vaccine development, such as ensuring optimal ribosome loading to maximize protein translation while avoiding ribosome clustering that could lead to mRNA degradation. The tool’s ability to safeguard against restriction enzyme cleavage sites and transcriptional terminators highlights its utility in designing sequences for experimental and therapeutic applications. These features align with the broader understanding of how codon adaptation influences mRNA stability and protein expression, making JCAT an invaluable resource for precise, biologically informed sequence optimization (Grote et al., 2005).
5 Secondary structure predictionPredicting the secondary structures of mRNA, including elements like α-helices and β-sheets, is essential for understanding its tertiary structure and function (Jiang et al., 2023). This prediction helps identify regions prone to degradation, allowing researchers to optimize gene sequences for greater mRNA stability. By analyzing the secondary structure, scientists can design mRNA sequences that are more efficient for translation, thereby improving vaccine expression in the host. Additionally, understanding the mRNA structure aids in selecting the most effective delivery systems, ensuring that mRNA efficiently enters cells and translates into target proteins, which enhances vaccine efficacy. Notably, CRISPR-Cas gene editing technology exemplifies its immense potential in addressing disease-causing mutations stemming from various cellular origins, highlighting the transformative impact of such evaluations on biomedical research and therapeutics (Cheng et al., 2020), and guiding the selection of those that can elicit stronger immune responses. In this section, we compare key tools for predicting mRNA secondary structure: RNAfold, mFold, and Inverse Prediction of RNA Knot (IPKnot).
5.1 RNAfoldRNAfold, part of the Vienna RNA Package, uses a thermodynamic model, such as the nearest neighbor thermodynamic model (Calonaci et al., 2020) to predict RNA secondary structures by computing the minimum free energy (MFE) and the thermodynamic regularized RNAfold can be used to calculate folding fractions that are highly correlated with the true free energy (Sato et al., 2021). RNAfold predicts RNA secondary structures by analyzing sequence inputs along with folding constraints, algorithms, and energy parameters. Users can select options for dangling ends, modified bases, and SHAPE reactivity data. The output can be customized to include interactive RNA secondary structure maps, reliability annotations, or mountain plots.
While RNAfold does not engage in direct codon optimization, its profound capability in predicting mRNA structures lays a solid foundation for subsequent codon optimization endeavors. Notably, key regions within mRNA, such as the 5’ UTR, 3’ UTR, and Poly(A) tail, play pivotal roles in facilitating vaccine translation, where the application of advanced techniques like sparsification can further enhance their efficacy (Gray et al., 2024), and RNAfold can assist in optimizing these regions to enhance vaccine expression levels. RNAfold also has several servers, such as RNAalifold, which can predict a set of common structures of aligned DNA or RNA sequences (Hofacker et al., 2002), which can calculate the hybridization energy and base pairing pattern of two RNA sequences (Bernhart et al., 2006). However, accurate RNA sequence data is essential for RNAfold’s predictions, and due to the complex diversity of RNA sequences, there may be some margin of error. Analyzing longer mRNA sequences also requires more computational time, which can significantly extend the development cycle.
5.2 MfoldMfold is a bioinformatics tool for predicting the secondary structure of RNA and DNA, similar to RNAfold. Similarly, Mfold contains several separate applications that can be used to predict nucleic acid folding, hybridization, and melting temperatures (Zuker, 2003). Mfold predicts the most likely secondary structure of a nucleic acid sequence by computing the most thermodynamically stable configuration (Zuker and Stiegler, 1981). This process involves calculating the free energy of various possible structures to determine which one is the most stable, which is essential for ensuring the stability of mRNA vaccines. Mfold uses dynamic programming algorithms to provide an optimal secondary structure based on the sequence and environmental conditions, such as Pknots-RE, NUPACK, gfold, and Knotty (Marchand et al., 2023) and the user can also change the rotation Angle to get the desired molecular folding orientation (Zuker, 2003). Unlike RNAfold, Mfold can identify regions in mRNA that might be prone to instability (Binet et al., 2023), such as regions with a high likelihood of forming secondary structures that may lead to degradation or poor folding. This sophisticated functionality empowers researchers to refine mRNA sequences with heightened precision for vaccine design, ensuring optimal performance. Furthermore, Mfold’s unique capability to anticipate the intricate interplay between mRNA molecules and delivery systems, and to visualize these interactions in various graphic formats including PostScript, PNG, or JPG, further augments its value in the realm of vaccine development (Zuker, 2003). Figure 2 presents a detailed comparison between RNAFold and Mfold predictions for mRNA secondary structure. As shown in Figure 2A, RNAFold provides a comprehensive visualization, with a color-coded structure based on base-pairing probabilities. Warmer colors highlight highly stable regions, particularly within the UTRs and near the poly-A tail—key areas for mRNA stability and translational efficiency. This allows for an in-depth understanding of structural stability across the mRNA sequence. In contrast, Mfold offers a simpler structural model without color-coding or probabilistic information. While it generates quicker results, Mfold’s predictions lack the depth required for a thorough stability analysis.
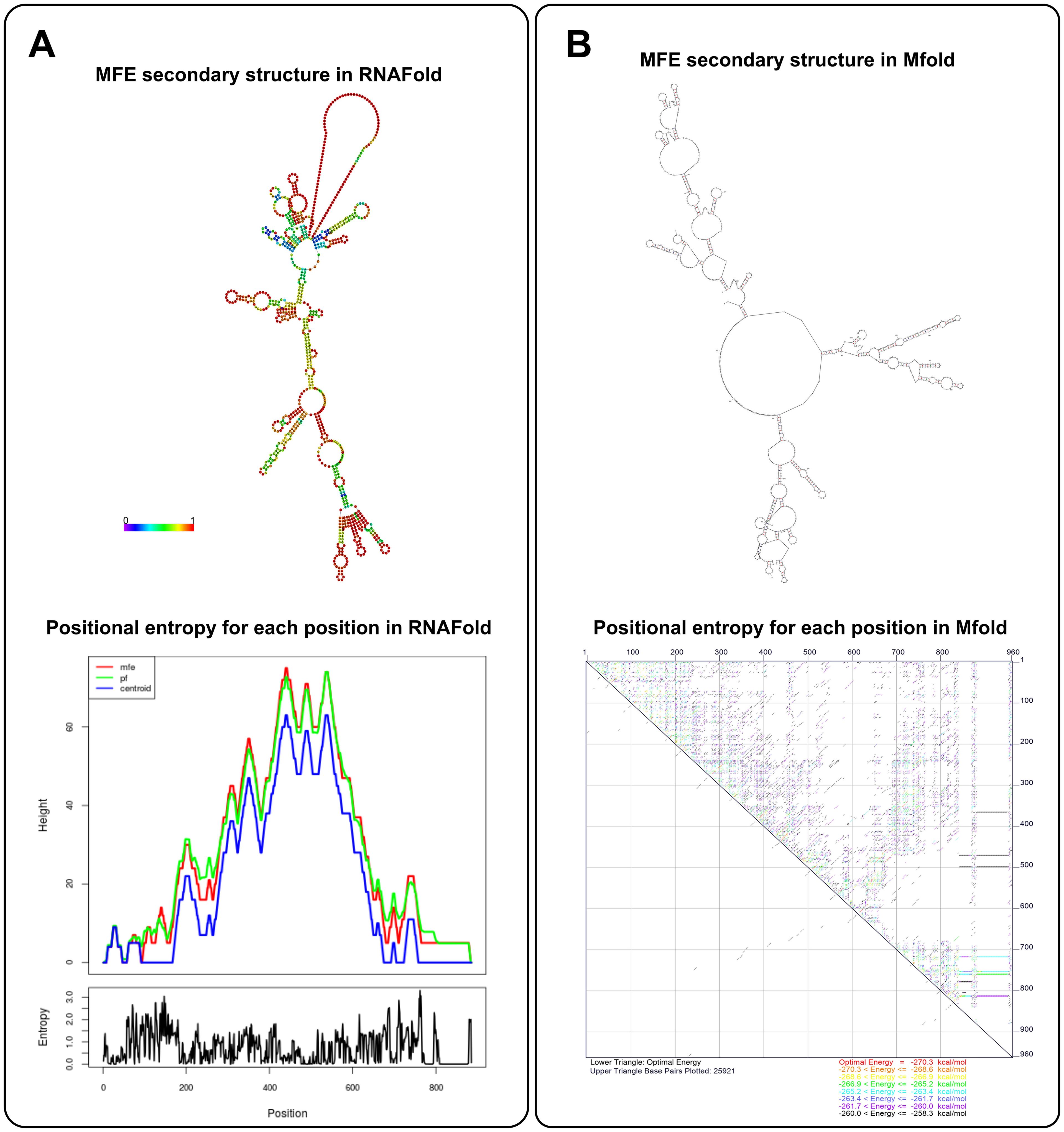
Figure 2. mRNA secondary structure prediction by RNAfold and Mfold. (A) The MFE structures for the template mRNA predicted by RNAFold and Mfold are shown. RNAFold (left) provides a detailed structural prediction with base-pairing probabilities, emphasizing stable regions-particularly in the UTRs and near the poly-A tail. This helps assess folding stability, essential for efficient translation. Mfold (right), while simpler, offers faster predictions, making it useful for quick structural overviews. (B) RNAFold’s positional entropy analysis shows low entropy in the UTRs and poly-A tail, confirming greater structural stability in these regions. Mfold provides a broader structural view but lacks detailed entropy data, making RNAFold more precise for stability assessment, while Mfold excels in speed and simplicity. The template used is a 962 bp mRNA encoding 12 neoantigens, with key regulatory elements like the HGH 5′ UTR, AES- mtRNA1-3′ UTR, and a 121-base pair long poly-A tail, designed for stability and efficient translation. The predictions were generated using RNAFold (ViennaRNA Package 2.4.18) and Mfold (version 3.6) (Zuker and Stiegler, 1981), with both tools sourced from their respective official repositories.
RNAFold further enhances its predictions with a detailed entropy analysis, using overlapping curves for MFE, probable folding pathways, and Centroid structures to illustrate structural variability at each nucleotide (Figure 2B). This analysis confirms low entropy in regions like the UTRs and poly-A tail, indicating stability in these essential areas. Although Mfold provides an energy-based prediction, it does not offer the same clarity in entropy distribution. As a result, RNAFold’s combination of structural and stability data makes it better suited for precise applications, while Mfold remains useful for rapid, less detailed evaluations.
5.3 IPKnotIPKnot is a specialized computational tool used to predict the secondary structure of mRNA molecules, providing critical insights into the folding process based on dynamic programming and thermodynamic principles (Kato et al., 2012). By simulating base-pairing interactions, IPKnot predicts structures such as hairpins, loops, and stems, which play a significant role in mRNA stability, translation initiation, and susceptibility to degradation by ribonucleases (Sato et al., 2011).
In the context of mRNA-LNP delivery, IPKnot’s folding predictions are essential for optimizing the interaction between mRNA and LNPs. The predicted mRNA secondary structure influences the mRNA’s ability to be encapsulated into LNPs, as well as the subsequent release and translation inside the target cell. IPKnot aids in designing mRNA sequences with secondary structures that are compatible with LNP formulations, enhancing encapsulation efficiency and promoting stable, controlled release into the cytoplasm. This stability is vital for maintaining the functional integrity of mRNA once inside the cell, ensuring that it can be efficiently translated to produce the encoded protein (Jabbari and Condon, 2014).
In cancer immunotherapy, specifically mRNA-based cancer vaccines, IPKnot plays a pivotal role in optimizing the mRNA sequence and its secondary structure for enhanced immune system activation. The folding pattern of the mRNA influences the conformation of the encoded antigen in MHC (Solheim et al., 1995). Efficient MHC class I and class II presentation is critical for triggering both CD8+ cytotoxic T cell responses and B cell-mediated antibody production against tumor-associated antigens. By fine-tuning the mRNA sequence to achieve an optimal secondary structure, IPKnot contributes to more efficient antigen presentation, thereby improving the activation of both the innate and adaptive immune systems. This leads to stronger and more sustained immune responses, which are essential for targeting and eradicating tumor cells (Bell et al., 2017). Moreover, IPKnot’s role in optimizing mRNA folding extends to improving the translational efficiency of mRNA in clinical applications, including gene therapies and personalized vaccines. The tool is integral in ensuring that mRNA molecules remain stable during synthesis, storage, and delivery, providing a foundation for the development of mRNA-based therapies with high efficacy and minimal degradation (Lee et al., 2024b). This capability is particularly crucial in the design of mRNA vaccines, where the accurate prediction of secondary structures ensures that the mRNA sequences are robust and capable of eliciting the desired immune response (Bon et al., 2008).
6 Protein structure predictionProtein structure prediction is crucial for understanding how mRNA vaccines generate their target antigens and interact within the host. Unlike costly proteomics techniques like gas chromatography-mass spectrometry (GC-MS), which analyze chemical compounds but don’t directly reveal protein structures, protein structure prediction provides theoretical insights crucial for refining antigen design before empirical testing. Accurate predictions ensure proteins fold correctly and function as intended, enhancing immune response. Key methods in this field include AlphaFold and Rosetta (Genc and McGuffin, 2025), which help identify potential folding and stability issues early, guiding experimental strategies and reducing extensive laboratory testing. Figure 3 compares mRNA structure predictions from AlphaFold and Rosetta.
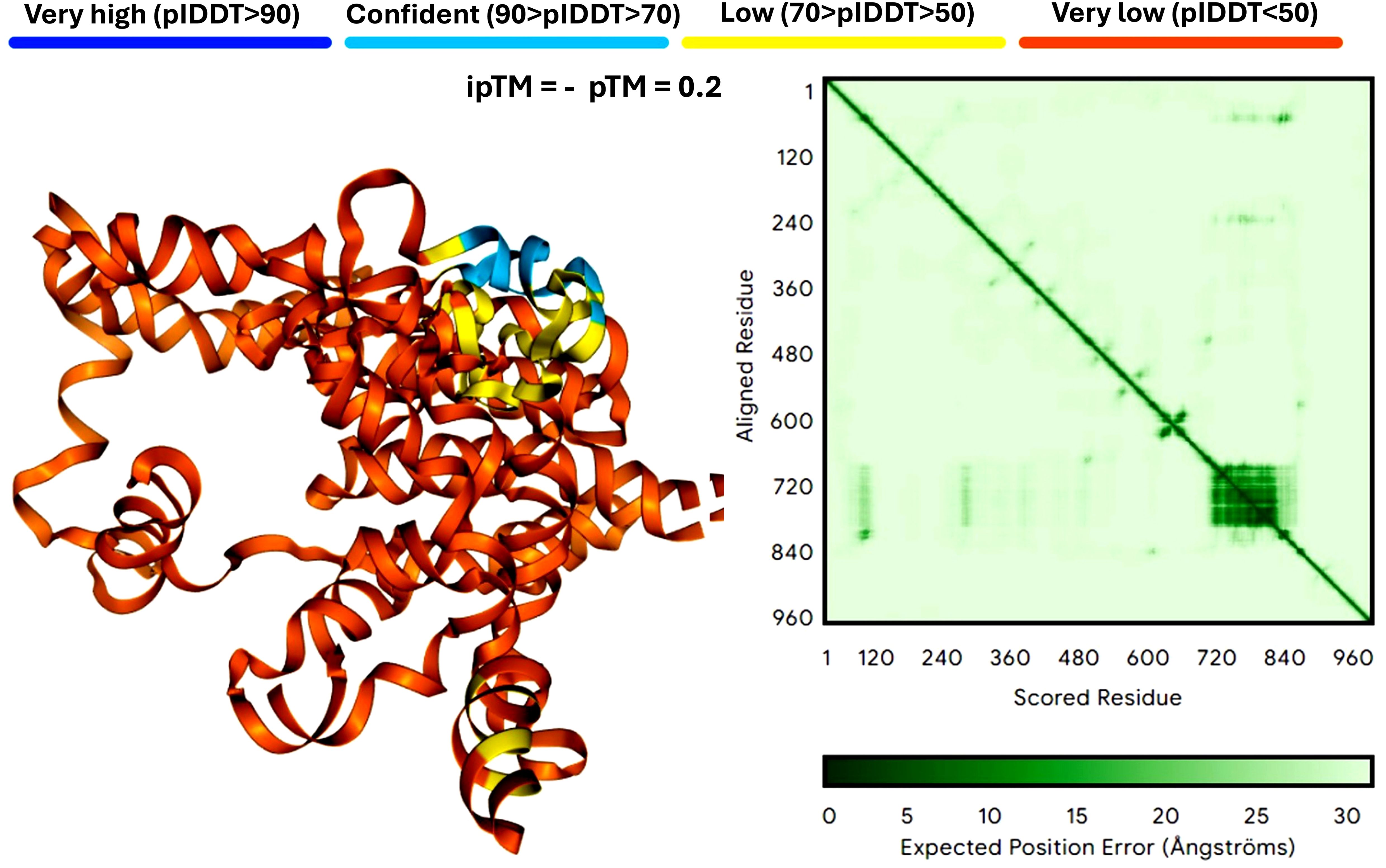
Figure 3. Comparison of mRNA structure predictions by AlphaFold. This figure illustrates the predicted three-dimensional structures of proteins derived from mRNA sequences, showcasing the strengths and limitations of AlphaFold. Panel A demonstrates AlphaFold’s capacity for detailed and accurate predictions for small to medium-sized proteins, while also highlighting its versatility in accommodating larger and more complex structures. The analysis is based on twelve neoantigens, featuring a YYA linker designed to enhance stability and facilitate efficient translation. The figure is powered by the latest version of AlphaFold 3 (accessible at https://alphafoldserver.com/) (Abramson et al., 2024). AlphaFold 3 is a web service that generates highly accurate biomolecular structure predictions for proteins, DNA, RNA, ligands, ions, and models chemical modifications for proteins and nucleic acids.
Protein structure prediction is essential for understanding how mRNA vaccines produce their intended antigens and how these proteins interact within the host. Distinct from intricate and expensive proteomics approaches, such as GC-MS, which delve into chemical compounds yet fall short in directly illuminating protein structures, protein structure prediction stands as a theoretical cornerstone for refining antigen design before empirical validation. Its precision is paramount, as it ensures that synthesized proteins adopt their correct folds and execute their intended functions, thereby fostering a potent immune response. Key methods, such as AlphaFold and Rosetta (Genc and McGuffin, 2025), are commonly used in this field. These approaches help identify potential issues in protein folding and stability early in the development process, guiding more effective experimental strategies and reducing the need for extensive laboratory testing.
6.1 AlphaFoldAlphaFold leverages deep learning techniques, such as the Attention Mechanism and Evolutionary Coupling algorithms, to predict the intricate 3D architectures of proteins from their amino acid sequences with atomic-level precision, even in the absence of prior structural knowledge (Jumper et al., 2021). Figure 3 shows the results of mRNA structure predictions by AlphaFold. In particular, AlphaFold can handle the missing physical environment and generate accurate models in challenging situations, such as intertwined homologs or proteins that fold only in the presence of an unknown heme group (Abramson et al., 2024). At the same time, AlphaFold has greatly improved the accuracy of structure prediction by combining a novel neural network architecture and training program based on evolutionary, physical, and geometric constraints of protein structure (Jumper et al., 2021). It combines a novel neural network architecture and training program rooted in evolutionary, physical, and geometric constraints to achieve unparalleled accuracy. With a database exceeding 214 million predicted protein structures, AlphaFold has transformed structural biology and set new benchmarks in protein structure prediction (Varadi et al., 2022; Abramson et al., 2024; Varadi et al., 2024), which are crucial for designing vaccines. However, it demands significant computational power and is dependent on the quality of input data. It works well for small to medium-sized proteins but may struggle with very large or complex proteins due to these resource limitations. DeepMind’s AlphaFold has set a new benchmark in protein structure prediction, employing advanced deep learning frameworks such as CNNs and attention mechanisms to achieve unparalleled precision. With a database containing over 214 million predicted protein structures, AlphaFold has profoundly influenced structural biology, providing a median backbone accuracy of 0.96 Å r.m.s.d.95 and an all-atom accuracy of 1.5 Å r.m.s.d.95 (Jumper et al., 2021).
In mRNA vaccine development, AlphaFold’s detailed structural insights are pivotal for optimizing antigen design, ensuring their stability and immunogenicity. Its ability to model viral proteins encoded by mRNA is crucial for assessing antigenicity, which is essential for effective vaccine formulations (Asediya et al., 2024). Furthermore, AlphaFold plays a significant role in enhancing the development of mRNA-LNPs delivery systems (2020;Asediya et al., 2024). LNPs encapsulate and protect mRNA during systemic circulation, facilitating targeted delivery and efficient intracellular release. AlphaFold’s structural predictions can guide the design of mRNA sequences with stable secondary structures, such as stem-loops or pseudoknots, to improve binding affinity and stability within LNPs. This optimization enhances protection against nuclease degradation and ensures efficient delivery to target cells (2020).
Additionally, AlphaFold can refine mRNA designs to optimize release kinetics from LNPs within target cells. By predicting how mRNA structures interact with LNP components in response to intracellular conditions, such as pH or enzymatic activity, AlphaFold aids in developing formulations that promote efficient unpacking and robust antigen translation upon endosomal escape. These advances enhance DCs activation and antigen presentation to T cells, ensuring a potent adaptive immune response (Jumper et al., 2021; Olawade et al., 2024). AlphaFold also supports the design of mRNA elements encoding immune-stimulatory adjuvants, amplifying immunogenicity when combined with LNP formulations. By optimizing antigen stability and presentation by MHC molecules, AlphaFold contributes to tailored mRNA-LNP formulations that elicit durable and specific immune responses. This capability is particularly valuable in cancer immunotherapy, enabling more precise and effective vaccine designs (Oladipo et al., 2024).
6.2 RosettaRosetta has become one of the leading computational tools for biomolecular structure prediction and design, using energy-based models, including Monte Carlo simulations and the Rosetta Energy Function (Rohl et al., 2004; Varadi et al., 2024), to predict 3D protein structures from mRNA sequences (Koehler Leman and Künze, 2023). It is useful for analyzing protein structures and interactions, and can also model antibodies and antigens (Schoeder et al., 2021), Rosetta can read most glycans in PDB files and automatically detect and score them, helping in the design of mRNA sequences to elicit strong immune responses. Rosetta provides detailed and adaptable predictions for proteins of various sizes, from small peptides to larger proteins, between 10-1000 residues (Du et al., 2021; Schmitz et al., 2021). However, it requires careful setup and parameter tuning, and its accuracy can decrease for very large or complex proteins.
7 Molecular dynamics simulationsMD simulations are crucial for understanding the intricate movements and interactions of atoms in mRNA molecules, especially how they interact with proteins and other cellular components (Hollingsworth and Dror, 2018). These simulations rely on several key parameters: Force fields are used to define atomic interactions, while temperature and pressure controls mimic physiological conditions. The permeability of lipid membranes is also considered to model interactions accurately (Venable et al., 2019). Time steps in simulations balance accuracy with computational efficiency. Cutoff distances manage non-bonded interactions, and periodic boundary conditions help minimize edge effects, enhancing the realism of the model. Solvent models simulate the surrounding aqueous environment, providing a more comprehensive view of mRNA behavior. Electrostatic treatments handle long-range interactions, ensuring that all aspects of the mR
留言 (0)