Acquiring and gathering clinical samples
The gathering of all cases from clinical samples involved in this study strictly adhered to the ethical guidelines of the Helsinki Declaration. All patients involved in the study have been fully informed about the research's purpose, methods, potential risks, and benefits and have provided written consent to participate. The research protocol has been submitted to the Institutional Review Board for review and has received formal approval (Approval number: No. TJ-IRB20220609).
The IVD tissue samples used in this investigation were sourced from patients experiencing pain or disability associated with IDD or undergoing other related treatments in our hospital from January 2019 to January 2023. These samples were specifically based on three key components of the disc: the annulus fibrosus (AF), nucleus pulposus (NP), and endplate (EP). Among them, 12 cases (average = 6, IDD = 6) were used for high-throughput transcriptome sequencing, and 20 cases (average = 10, IDD = 10) were used for immunohistochemical staining to validate the patients, Table S1 for more details (Li et al. 2022a, b).
Transcriptome sequencing
Extract total RNA from each sample employing Trizol reagent (Thermo, 16096020, USA). Assessment of RNA concentration, purity, and structural integrity involved the Qubit® 2.0 Fluorometer® (Life Technologies, Q33216, USA), NanoPhotometer (IMPLEN, USA), and the RNA Nano 6000 Assay Kit on the Bioanalyzer 2100 system (Agilent, 5067–1511, USA). The input material for RNA sample preparation is 3 μg of total RNA in every sample. In line with the instructions from the producer, cDNA libraries were synthesized utilizing the NEBNext® UltraTM RNA Library Prep Kit (NEB, E7435L, Beijing, China) specifically designed for Illumina® sequencing, and the quality assessment was carried out with the Agilent Bioanalyzer 2100 system. The clustering of indexed-encoded samples on the cBot cluster generation system was carried out as per the instructions provided by the manufacturer for the TruSeq PE Cluster Kit v3 cBot HS (Illumina, PE-401–3001, USA). Subsequent to the clustering step, the library underwent sequencing utilizing the Illumina-Hiseq 550 platform, yielding paired-end reads of 125 bp/150 bp in length.
The quality verification of paired-end reads from the raw sequencing data was executed through the application of FastQC software version 0.11.8. Raw data preprocessing encompassed the removal of Illumina sequencing adapters and poly(A) tail sequences utilizing Cutadapt software version 1.18. Remove reads with N content over 5% by perl script. The FASTX Toolkit software version 0.0.13 facilitated the extraction of 70% of high-quality bases from the sequencing reads, based on a threshold score of 20. The utilization of BBMap software was instrumental in rectifying the paired-end sequences. The processed superior reads were matched against the human reference genome employing the hisat2 tool (version 0.7.12) (Arunachalam et al. 2022; Linkner et al. 2021; Deng et al. 2020; Peng et al. 2019).
MSI-IDD mouse modeling and treatment
Approval for the experimental procedures and animal usage protocols was granted by the Animal Ethics Committee at our hospital under the designation TJH-202107013. The animal trials undertaken in this research were compliant with internationally recognized animal welfare standards and relevant regulations. Every possible measure was taken during the experiments to minimize animal pain and discomfort. At the conclusion of the experiments, the animals were euthanized in a humane manner.
Forty-six 12-week-old male C57BL/6J mice weighing 20 ± 2 g were sourced from Beijing Vitonlihua Experimental Animal Technology Co., Ltd. (219, Beijing). Housing arrangements for the mice involved placing them in standard feeding cages within an environment set at a consistent room temperature of 23 ± 1°C, following a 12–12 h light–dark cycle, and ensuring continuous access to food and water. Acclimation period of one week was provided for the mice before initiating the experiments.
The msi-IDD model was established by applying a constant torque. After anesthetizing the mice, their tails were cleaned with 70% ethanol. The IVD space between the 9th and 10th caudal vertebrae was located by palpation, and the corresponding area on the skin was marked. Using a drill, two stainless steel pins with a diameter of 0.4 mm were percutaneously inserted into the vertebrae adjacent to the marked disc, positioning them centrally within the vertebral body and perpendicular to the tail axis. After seven days, a 1.4 mm thick elastic band was calibrated to exert a compressive force of 1.7 N at a distance of 2.5 mm from the disc center and was placed between the two pins on the left side of the tail. A stainless steel tube was pressed onto the pins to secure the elastic band’s position. Additionally, to prevent the mice from damaging the elastic band or being injured by the pins, a plastic shield was fixed to the proximal pin with a hook (Daly et al. 2016). In the sham-operated group (Control), stainless steel pins were implanted, but the elastic band was not applied. After seven days of bending, the protective shield and elastic band were carefully removed, and further experiments were conducted.
After the model was established, IVD tissue samples (C2-C7; based on the AF, NP, and EP) were collected from 3 Control and 3 msi-IDD mice for single-cell sequencing.
The mice were randomly divided into four groups of 10 each: Control, msi-IDD, msi-IDD + sh-SPP1 + Mock, and msi-IDD + sh-SPP1 + GSK2656157. The sh-SPP1 was delivered using an adeno-associated virus (AAV5) vector, with virus packaging services provided by Sangon Biotech (Shanghai, China). AAV was injected into the Co9/10 disc using a 33G needle (1 × 1012 GC/mL in 2 μL saline) every two weeks. For drug treatment, GSK2656157 (0.5 mg/kg, MCE, HY-13820) was injected into the Co9/10 disc using a 33G needle, while the control group received an equivalent volume of saline, administered three times per week for four weeks (Teng et al. 2023). At the end of the experiment, the mice were euthanized by CO₂ inhalation, and relevant tissues were collected as needed for further experiments.
Single-cell transcriptome sequencing of mouse IVD tissue
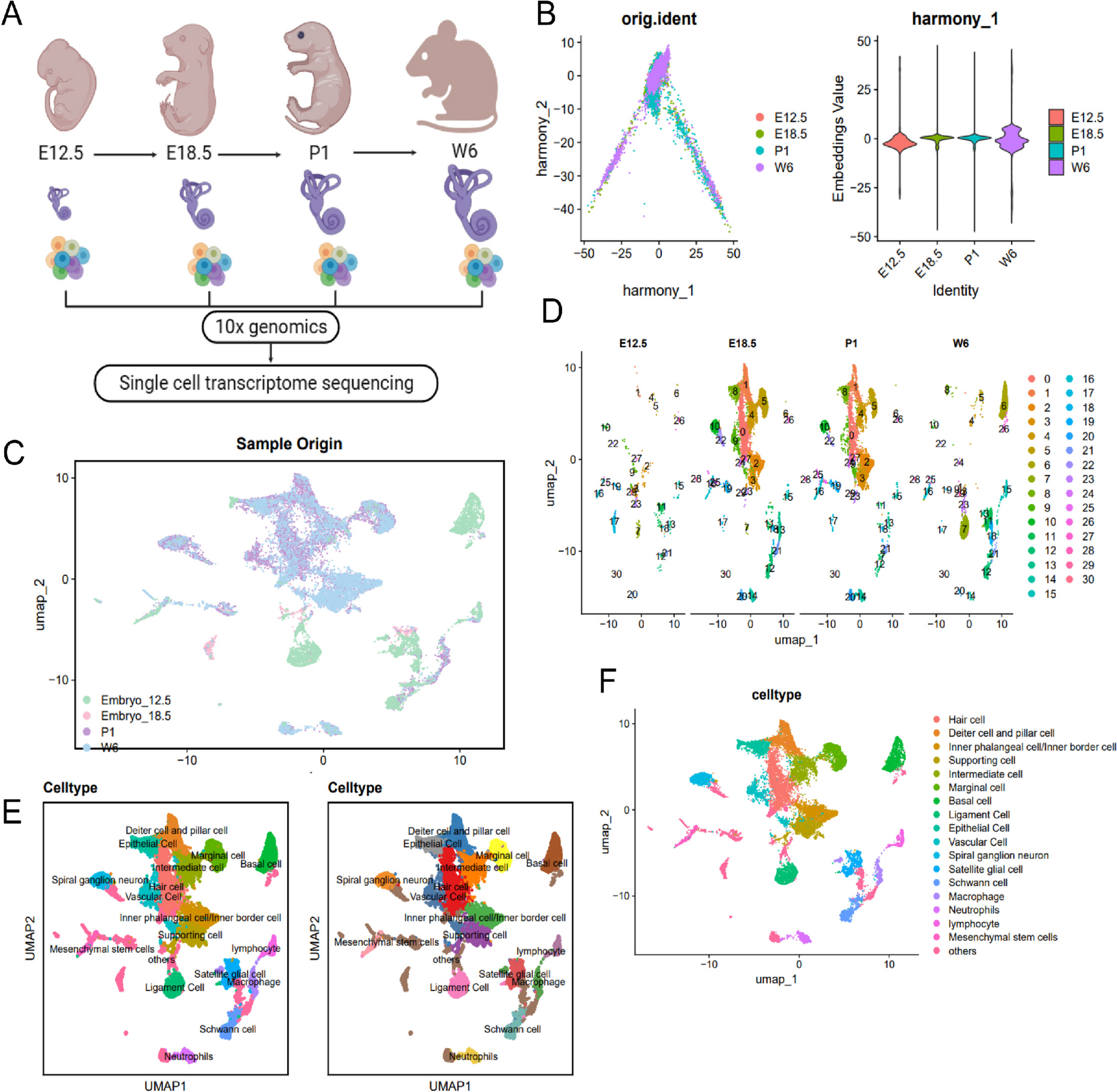
First, the obtained IVD tissue (C2-C7; AF, NP, and EP) was washed with cold PBS to remove excess tissue and dissociated. The tissue was then digested with 1 mg/mL collagenase (C2674, Sigma-Aldrich, USA) at 37°C for 10 min, followed by an additional 5-min incubation with trypsin/EDTA (25200072, Gibco, USA) to prepare a single-cell suspension. Single cells were captured using the C1 Single-Cell AutoPrep System (Fluidigm, Inc., South San Francisco, California, USA) and lysed within the chip, releasing mRNA and synthesizing cDNA through reverse transcription. The lysed and reverse-transcribed cDNA was pre-amplified in a microfluidic apparatus for upcoming sequencing. The cDNA that underwent amplification was used for library construction and sequenced on the HiSeq 4000 Illumina platform (utilizing paired-end reads, with a read length of 2 × 75 base pairs, generating roughly 20,000 reads per cell) (Keefe et al. 2023). Data quality control was performed using the criteria of nFeature_RNA > 200 & nCount_RNA < 20000 & percent.mt < 1, and a subset of 2000 genes displaying significant expression variability was identified (Olaniru et al. 2023). The single-cell RNA sequencing (scRNA-Seq) dataset's dimensionality was reduced through principal component analysis (PCA) that concentrated on the top 2000 genes with the most significant expression variability. The Elbowplot function within the Seurat package was utilized to select the initial 17 principal components for downstream analysis purposes. Utilizing the resolution parameter of 0.5, the FindClusters function in Seurat was employed to discern the main cellular subtypes. Subsequently, the scRNA-seq data was nonlinearly reduced using the UMAP algorithm. Finally, known cell lineage-specific marker genes were utilized, along with annotation from the online resource CellMarker, for cellular annotation (Table S2) (Shao et al. 2020).
Maintenance of neural cells and macrophage progenitors cell lines
The RAW 264.7 mouse macrophage cell line (TIB-71, USA) obtained from ATCC was cultured in Thermo Fisher's DMEM-H (11965092, USA). The THP-1 human peripheral blood monocyte cell line (CRL-3304, USA) provided by ATCC was cultured in Thermo Fisher's RPMI 1640 (11875119, USA). The mouse nucleus pulposus cells (mNPCs) (CP-M146, China, provided by Procell) were cultured in Thermo Fisher's DMEM/F-12 (11320033, USA). In all cell culture processes, a mixture of 10% FBS (10100147C, Thermo Fisher, USA) and 1% Penicillin–Streptomycin (15140163, Thermo Fisher, USA) was supplemented, and the cellular incubation was conducted at 37°C under 5% CO2 atmosphere (Jiang et al. 2011). The differentiation of THP-1 cells led to the development of macrophages by treating them with 200 nM PMA for 72 h at a seeding density of 0.2 × 106 cells/ml.
To validate the SPP1/PERK signaling axis and its impact on macrophage secretory phenotype and msi-mNPCs, CCT020312 (5 μM, HY-119240, USA) provided by MedChemExpress was used to activate PERK, or GSK2656157 (20 nM, HY-13820, USA) was used to inhibit PERK (Bruch et al. 2017). The subsequent experiments were carried out after 48 h of cell treatment with the drugs. The mentioned cell experiments were duplicated in triplicate as part of the research protocol.
Lentiviral infection
The following cell lines were constructed using lentivirus-mediated transfection: SPP1 overexpression (oe-NC and oe-SPP1) and knockdown (sh-NC and sh-SPP1) cell lines based on RAW 264.7 cells, along with their controls; ATF4 overexpression (oe-NC-RAW and oe-ATF4-RAW) and knockdown (sh-NC-RAW and sh-ATF4-RAW) cell lines based on RAW 264.7 cells, along with their controls; and ATF4 overexpression (oe-NC and oe-ATF4) and knockdown (sh-NC and sh-ATF4) cell lines based on THP-1 cells, along with their controls. Knockdown cell lines were constructed using two shRNA sequences, with the sequence showing higher knockdown efficiency selected for subsequent experiments. The shRNA interference sequences are provided in Table S3.
The plasmids and lentiviral packaging services were provided by Sangon Biotech (Shanghai, China). The constructed plasmids carrying the target gene and auxiliary plasmids were co-transfected into 293T cells (CRL-3216, ATCC, USA). After verification, amplification, and purification, packaged lentivirus was obtained. For lentivirus-mediated cell transfection, 5 × 105 cells were seeded into a 6-well plate. When cell confluence reached 70–90%, an appropriate amount of packaged lentivirus (MOI = 10, working titer approximately 5 × 106 TU/mL) and 5 μg/mL polybrene (TR-1003, Merck, USA) were added to the culture medium for transfection. After 4 h, an equal amount of culture medium was added to dilute the polybrene, and the medium was replaced with fresh medium after 24 h. Transfection efficiency was assessed 48 h later using a luciferase reporter gene, and stably transfected cell lines were selected using 1 μg/mL puromycin (A1113803, Thermo Fisher, USA) (Kumar et al. 2018).
Construction of mNPCs mechanical stress damage model
Once the NPC confluence attained 80%, cell detachment was achieved through digestion using 0.25% trypsin–EDTA, then the cells were suspended in a culture medium mix consisting of DMEM/F-12 and 4% low melting point agarose. The mixed solution is incubated in a 6-well BioFlex plate. Compression trials were executed employing the Flexcell FX5000 device (Flexcell International, McKeesport, USA). The frequency parameter is configured as 1.0 Hz, the compression intensity is 15%, and the cells are collected after 48 h of treatment. The control group is nurtured in similar environments, yet devoid of mechanical strain. The injury condition is assessed using flow cytometry and ELISA (Shi et al. 2022).
The phenotypic effect of macrophages on msi-mNPCs was assessed using a co-culture method. The experiment was performed using a 0.4 μm Transwell, with the seeding of msi-mNPCs in the upper chamber at a concentration of 105 cells per well. To verify the impact of macrophage SPP1 expression on msi-mNPCs, the lower chamber was sequentially seeded with the control group (Transwell upper layer with culture medium only, no cells, referred to as Mock), oe-NC, and oe-SPP1; or Mock, sh-NC, and sh-SPP1. To investigate the influence of the macrophage SPP1/PERK signaling axis on msi-mNPCs, the lower chamber was sequentially seeded with sh-SPP1 + Mock, sh-SPP1 + GSK2656157 (post-treated with GSK2656157); or sh-SPP1 + sh-NC-ATF4, sh-SPP1 + sh-ATF4; or sh-SPP1 + Isotype, sh-SPP1 + anti-IL-10. After 48 h of co-culture, msi-mNPCs were harvested for subsequent experiments (Shi et al. 2022).
The upper chamber of the Transwell insert was filled with msi-mNPCs, while the lower chamber was inoculated with oe-PERK-M2, and anti-IL-10 (10 μg/mL, Bio X cell, BE0049, USA) was supplemented in the lower chamber as per experimental requirements. After 48 h of co-culture, msi-NPCs were harvested for further experiments (Tai et al. 2013).
ChIP, DNA-Pull down, and dual-luciferase experiments
Once the cell fusion reached 70–80%, 1% formaldehyde was introduced at room temperature to stabilize the DNA–protein complexes within the cells for 10 min. Following fixation, the complexes were randomly disrupted using ultrasonication for 10 s with a 10-s pause between each cycle, totaling 15 cycles for the fragmentation into suitably sized pieces. The lysate was subject to centrifugation at 12,000 g at 4°C. The supernatant was divided into two tubes and incubated separately with a negative control antibody, IgG (1 μg/ml, Abcam, ab171870, UK), and the specific antibody against ATF4 (1 μg/ml, Abcam, ab85049, UK) overnight at 4°C. The native DNA–protein assemblies were extracted by employing Protein Agarose/Sepharose for precipitation, and subsequent to rapid centrifugation, the excess fluid was disposed of. The nonspecific complexes were washed, and the cross-linking was reversed by overnight exposure to 65°C. Purification of the DNA fragments was achieved by employing phenol/chloroform extraction. The ChIP-PCR products were characterized by 3% agarose gel electrophoresis (Yang et al. 2016).
THP-1 cells were transfected with 50 nM biotin-labeled Biotin-IL-10-Wt (5'-AGGTGATGTAATAT-3') / Biotin-IL-10-Mut (5'-ACCACTACTTTATT-3') (Goldengate, Wuhan, China) and collected post 2-day duration. The cells were cleansed with PBS solution. After cell lysis, the lysate was mixed with M-280 streptavidin magnetic beads pre-coated with BSA and yeast tRNA without RNase (Merck, 55714, USA), followed by a 3-h incubation at 4°C with constant agitation. The cell lysate underwent a dual washing process with pre-chilled lysis buffer, proceeded by three rounds of washing with buffer solution containing minimal salt content, and finalized with a single wash using high salt buffer. The enrichment of the relevant proteins was detected using Western blot analysis (Luan et al. 2018).
Based on the JASPAR database prediction of the binding sites between ATF4 and the IL-10 promoter region, wild-type (Wt, 5'-AGGTGATGTAATAT-3') and mutant-type (Mut, 5'-ACCACTACTTTATT-3') sequences of IL-10 were constructed. Incorporation of these sequences into the pGL-3 luciferase reporter vector (Thermo Fisher, 4351372, USA) was carried out. The luciferase reporter plasmids were transfected into oe-NC or oe-ATF4 cells constructed based on THP-1 cells. After 48 h, cell collection was followed by lysis via centrifugation at 250 g for a duration of 3 to 5 min. After harvesting the supernatant, the luciferase activity was quantified with the Dual-Luciferase® Reporter Assay System (Promega, E1910, USA). Firefly luciferase functionality was determined by the introduction of 100 μL of Firefly luciferase working solution into the cell samples, while measurement of Renilla luciferase activity involved the utilization of 100 μL of Renilla luciferase working solution. Evaluation of the relative luciferase activity was achieved by assessing the Firefly luciferase to Renilla luciferase activity ratio (Jin et al. 2020). The test was duplicated thrice for validation.
Detection of mNPC proliferation and apoptosis
The CCK-8 assay kit (BeyoSky, C0037, Shanghai, China) detects cell proliferation. Cultured cells in the logarithmic growth phase were maintained in DMEM/F-12 medium with a 10% concentration of FBS to adjust the concentration to 5 × 104 cells/mL. Then, they were distributed into a 96-well culture plate, with 100 μL of cell suspension dispensed into each well. Incubation lasting 48 h occurred in a CO2 incubator after the plate was situated. Subsequently, 10 μL of CCK-8 solution was applied to individual wells after discarding the supernatant swiftly. After incubation at 37°C for 2 h, the measurement of absorbance at 450 nm was conducted utilizing a Multiskan FC microplate reader (Thermo Fisher, 51119080, USA) (A). Set 3 parallel wells in each group and take the average value.
Perform apoptotic cell detection by flow cytometry utilizing the Pacific Blue™ Annexin V/SYTOX™ AADvanced™ Apoptosis Detection Kit (Thermo, A35136, USA), complying with the manufacturer's directives (Wu et al. 2021).
Immunohistochemistry
Samples from IVDs were preserved with 4% paraformaldehyde, treated with EDTA for decalcification, and subsequently enclosed in paraffin to enable tissue sectioning. The technique of immunohistochemistry was applied to explore the expression and distribution of relevant proteins in the IVD tissue. Subsequent to the rehydration step for the sections, antigen retrieval was executed following the recommended procedure for the primary antibodies. Afterward, the staining process proceeded with the application of a universal two-step staining kit (Zhongshan Golden Bridge, PV-9000, Beijing, China), which included all reagents except the primary antibodies. The primary antibodies used in this study were as delineated below: anti-CD11b (human/mouse, 1:4000, Abcam, ab133357, UK), anti-Collagen Iα1 (mouse, 1:500, Abcam, ab270993, UK), anti-Collagen II (mouse, 1:400, Abcam, ab34712, UK), anti-IL-1α (mouse, 1:500, Abcam, ab300499, UK), and anti-TNF-α (mouse, 2.5 μg/mL, Abcam, ab1793, UK).
The utilization of an optical microscope (Olympus, CX43, Japan) enabled the observation and preservation of the staining results. An analysis with a semi-quantitative approach was carried out on the outcomes employing Image Pro Plus software (Daniel et al. 2021).
Enzyme-linked immunosorbent assay (ELISA)
Cell culture supernatant (CM) was collected and samples were tested for the expression levels of IL-10 (Abcam, ab255729, UK), transforming growth factor beta 1 (TGF-β1, Abcam, ab119557, UK), MMP-13 (Huamei Biotech, CSB-E07413m, Wuhan, China), Collagen Iα1 (Abcam, ab210579, UK), Collagen II (Huamei Biotech, CSB-EL005739HU, Wuhan, China), Aggrecan (Abcam, ab213754, UK), IL-1α (ab113344), and TNF-α (Abcam, ab208348, UK) in the samples using ELISA reagent kits, complying with the directions outlined by the manufacturer (Hou et al. 2021).
IVD histological staining
The mouse IVD tissue was immersed in a 10% neutral formalin buffer for fixation, followed by embedding and slicing. For the organization of H&E staining, the tissue sections were swiftly cleansed with 1 × PBS for a mere 2 s, stained with Hematoxylin for 60 s at 60°C, washed with 1 × PBS for 10 s, differentiated with 1% hydrochloric acid alcohol differentiation solution for 3 s, washed with 1 × PBS for 2 s, stained with Eosin for 3 min, washed with 1 × PBS for 2 s, dehydrated with 70%, 80%, 95% ethanol and absolute ethanol for 5 min respectively, cleared with xylene three times for 5 min each, and ultimately, the sections were enclosed with mounting medium. In the process of evaluating tissue organization through Safranin-O/Fast Green staining, after rehydration of the sections, stain with 0.1% Safranin-O solution for 5 min, followed by ethanol decolorization, stain with 0.1% Fast Green solution for 5 min, and finally dehydrate and mount the slides. The visualization and recording of the staining results were accomplished with the assistance of an optical microscope (Olympus, CX43, Japan), and utilization of the Image Pro Plus software enabled semi-quantitative evaluation of the outcomes. The histological scoring was conducted according to the Masuda criteria. The more elevated the score, the more pronounced the degenerative state (Teng et al. 2023).
Western blot
Total protein was derived from the samples utilizing the protein extraction kit (Bestbio, BB3101, Shanghai, China), while the protein concentration was quantified with the BCA assay kit (Beyotime, P0012S, Shanghai, China). Prepare a 10% SDS-PAGE gel (Bi Yuntian, P0012A, Shanghai, China). Add 50 μg of protein sample to each well. Perform constant voltage electrophoresis at 80 V for 2 h, followed by 120 V for 2 h. 250 mA constant current wet transfer for 90 min, transferring the proteins to the PVDF membrane (Merck, IPVH00010, Germany). Incubate the PVDF membrane at room temperature with TBST comprising 5% non-fat dry milk powder for 2 h, discard the blocking solution, and wash with TBST for 10 min. Incubate at 4 °C overnight with 1:1000 dilution (antibody information in Table S4). Perform triple washing in TBST, with each wash lasting 10 min. Goat anti-rabbit IgG conjugated with horseradish peroxidase (1:2000, Abcam, ab6721, UK) or Goat anti-mouse IgG (1:2000, Abcam, ab6789, UK) was incubated at ambient temperature for one hour, then rinsed thrice with PBST for 10 min per rinse. ECL reaction solution (Biyun Tian, P0018FS, Shanghai, China) is used for color development, followed by exposure and development in a dark containment. Threefold repetitions are implemented for every sample test (Chen et al. 2022).
RT-qPCR
Trizol (Thermo Fisher, 16096020, USA) was used for total RNA extraction from the samples, followed by reverse transcription and PCR (one-step) using the One Step TB Green® PrimeScript™ RT-PCR Kit (Takara, RR066A, Japan). The execution of the RT-qPCR reaction took place in the Thermal Cycler Dice™ Real-Time System III (Takara, TP990, Japan) platform. The program includes a reverse transcription phase (42 ℃ 5 min, 95 ℃ 10 s, cycle number = 1), a PCR phase (95 ℃ 5 s, 60 ℃ 34 s, cycle number = 40), and a melt curve phase (95 ℃ 15 s, 60 ℃ 1 min, 95 ℃ 15 s, cycle number = 1). After the reaction is complete, we confirm the amplification curve and the melting curve. By employing the 2−ΔΔCt approach, we ascertain the relative expression levels of the target gene between the experimental and control groups. In this method, ΔΔCt = ΔCt experimental group—ΔCt control group, and ΔCt = Ct target gene—Ct reference gene. The amplification number of cycles required for the reaction to reach the set threshold during amplification is denoted by Ct. We conducted three repetitions of the experiment; each time, the sample contained three duplicate wells. The primer sequences could be referred to. We used GAPDH as an internal reference (Tian et al. 2022).
Statistical analysis
Bioinformatics results were statistically analyzed using R version 4.3.0, and other results were analyzed using GraphPad Prism (San Diego, CA, USA). Quantitative data are presented as mean ± standard deviation. Normality and homogeneity of variances were first assessed; if both conditions were met, an independent t-test was used to compare differences between the two groups. For comparisons among three groups, a one-way analysis of variance (ANOVA) followed by Tukey's post hoc test was applied. Statistical significance was set at P < 0.05.







留言 (0)