
記住我






Compliance with ethical directives for the treatment and utilization of laboratory animals, encompassing not exclusively the "3R Principle" (Replacement, Reduction, Refinement), was followed in all animal tests. Authorization to conduct the experimental protocols in this study was granted by the Animal Ethics Committee of China Medical University (Ethical Review Approval Number: CMUXN2022723) to ensure respectful and humane treatment of all experimental animals.
Cell culture and treatmentShanghai Enzyme Research Biotechnology Co., Ltd. (Shanghai, China) provided the human THCA cells TPC-1 (CC-Y1522) and B-CPAP (CC-Y1064). Cellular cultures were maintained in DMEM medium rich in glucose (11965118, Thermo Scientific, USA), accompanied by 10% fetal calf serum (26140079, Thermo Scientific, USA) and 1% penicillin–streptomycin (100 U/mL penicillin and 100 μg/mL streptomycin) at 37 °C under 5% CO2 conditions using an incubator with humidity control (BB15, Thermo Scientific, USA).
Cell treatmentTo achieve CHEK1 overexpression in TPC-1 and B-CPAP cells, lentiviral infection was utilized. Sangon Biotech, situated in Shanghai, China, offered services for lentivirus packaging. Introduction of the vector pHAGE-puro series plasmid, along with the auxiliary plasmids pSPAX2 and pMD2.G, into 293 T cells (CL-0005, Procell Life Science & Technology Co., Wuhan, China) occurred through co-transfection. Supernatants were gathered, centrifuged, and concentrated post 48 and 72 h of cell culture. The viral supernatants were pooled, and titers were determined. In 6-well plates, cells were distributed at a concentration of 1 × 105 cells per well and were exposed to an appropriate dosage of lentiviral particles (MOI = 10, active concentration ~ 5 × 106 TU/mL) along with 5 μg/mL polybrene (Merck, TR-1003, USA) upon achieving 75% cell confluence. Post 4 h of infection, an equivalent quantity of medium was introduced to diminish the polybrene concentration, and 24 h later, fresh medium was substituted for the original medium used. For stable cell line construction, the selection of resistant cells was achieved through the use of puromycin (5 μg/mL, Sangon Biotech, A100339, Shanghai, China).
Identification of Ginsenosides-Rh2 target genesUtilizing the resources of the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), we generated chemical structure depictions of Ginsenosides Rh2 in both 2D and 3D formats (Figure S1). An SDF-formatted file containing the 3D depiction of the chemical structure was uploaded to the PharmMapper server. The PharmMapper reverse docking server was utilized to conduct molecular docking simulations for a variety of active compounds, with a specific focus on "Human Protein Targets Only" and default parameters, in order to pinpoint potential targets for Ginsenosides-Rh2.
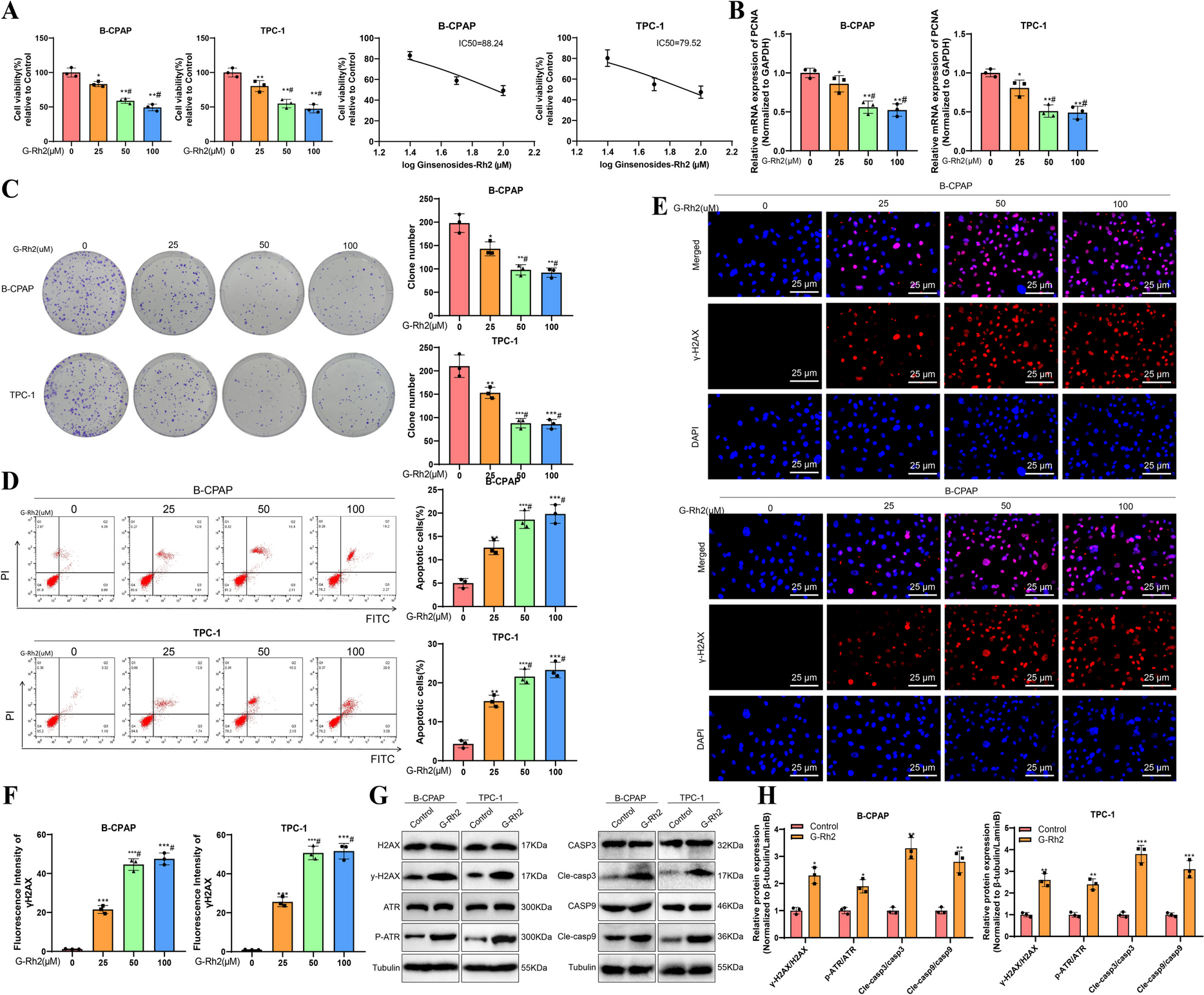
CCK-8 assayFor the CCK-8 assay, cell digestion occurred followed by resuspension at a concentration of 1 × 105 cells/mL, then establishment involved introducing 100 μL of the cellular composition into the wells of a 96-well plate, proceeding with an overnight incubation as prescribed. Cell viability was determined at the 48-h time point post-seeding in adherence to the instructions outlined in the CCK-8 assay kit (C0037, Beyotime, Shanghai, China). During every assessment, 10 μL of CCK-8 detecting solution was included, and subsequent to a 4-h incubation in the cell culture incubator, the plate was positioned for absorbance measurement at 450 nm with a microplate reader to generate a growth curve (Zhang et al. 2022).
Colony formation assayWhen performing the colony formation test, the cells were dispersed into singular cell suspensions, and then 1000 cells were distributed evenly into every 6 cm culture dish with a complete growth medium. After 48 h of incubation, a procedure involving PBS wash, fixation with 4% paraformaldehyde (PFA), and staining using 0.4% crystal violet for 15 min was executed on the cells (Wei et al. 2020). More than 10-cell colonies were manually enumerated, and the average count from three replicate wells was calculated.
Flow cytometry (FCM) analysis of apoptosisThe recognition of cell apoptosis was achieved utilizing the Annexin V-FITC/PI apoptosis detection kit (Cat. No: C1062M, Beyotime, Shanghai, China). Cell density of 1 × 106 cells per milliliter was used, undergoing two washes with chilled PBS. Subsequently, cells were softly suspended in 195 μL of binding buffer containing Annexin V-FITC, accompanied by 5 μL of Annexin V-FITC binding agent and 10 μL of propidium iodide. Utilizing a flow cytometer (FACSVerse, BD, USA), cell apoptosis quantification was executed following a 15-min dark incubation at room temperature (RT) (Wang et al. 2020a, b). Apoptotic cells were tallied in the upper right (UR) and lower right (LR) quadrants.
γ-H2AX immunofluorescenceCell fixation was achieved by applying 4% PFA for 20 min at RT, proceeded by 5 min of permeabilization using 0.1% Triton X-100. The cells were subsequently placed in culture overnight with primary antibody Anti-γH2AX (ab81299, 1:250, Abcam, UK) at 4 °C, followed by rinsing and subsequent exposure to secondary antibody goat anti-rabbit IgG H&L (Alexa Fluor® 555) (ab150078, 1:1000, Abcam, UK) for 1 h at RT. The cellular nuclei underwent DAPI staining (4 µg/ml, 62248, Thermo Scientific) for 30 min at RT. Observation and analysis of stained cells were performed with a confocal laser scanning microscope (Olympus Corporation) followed by processing in Image J software (Cai et al. 2021).
qRT-PCRThe isolation of RNA from tissues and cells involved the use of Trizol reagent (16096020, Thermo Fisher Scientific, USA). Assessment of RNA concentration and purity was carried out employing a NanoDrop One/OneC spectrophotometer by Thermo Scientific, with the goal of achieving a A260/A280 ratio of 2.0 and a concentration surpassing 5 μg/μL. The generation of cDNA was achieved by reverse-transcribing the RNA with the First Strand cDNA Synthesis Kit (D7168L, Beyotime, Shanghai, China). The RT-qPCR procedure, conducted with the Vazyme RT-qPCR kit (Q511-02, Nanjing, China), adhered to the operating guidelines set by the manufacturer. Sangon Biotech in Shanghai, China was responsible for creating and offering the primer sequences, which are detailed in Table S1. By applying the 2−ΔΔCt method, the levels of target genes were determined in the experimental and control groups, employing GAPDH as the reference gene. The formula applied was ΔΔCt = ΔCt (experimental group)—ΔCt (control group), where ΔCt = Ct (target gene)—Ct (reference gene) (Wang et al. 2020a, b). The trial was reiterated thrice for accuracy assessment.
Western blot (WB)The complete extraction of protein was accomplished by the addition of 1% PMSF to RIPA lysis buffer (P0013B, Beyotime, Shanghai) for cell lysis. NE-PER™ cytoplasmic and nuclear extraction reagents (78833, ThermoFisher Scientific, USA) were utilized to extract nuclear proteins, with subsequent measurement of the overall protein level in every sample applying a BCA assay kit (P0011, Beyotime, Shanghai). Subsequently, SDS gels with 8%−12% concentration were created based on the size of the target proteins, and even distribution of protein samples across each lane was ensured by employing a micropipette for the process of electrophoretic separation. The transition of proteins from the gel to a PVDF membrane (1620177, BIO-RAD, USA) occurred, and they were blocked with 5% skim milk for 1 h at RT.
Antibodies including anti-CHEK1 (ab32531, 1:1000), anti-pCHEK1 (ab79758, 1:1000), anti-H2AX (ab124781, 1:1000), anti-gamma H2AX (ab81299, 1:2000), anti-LaminB (ab16048, 1:1000), anti-ATR (ab2905, 1:1000), anti-p-ATR (ab178407, 1:1000), anti-caspase3 (ab32351, 1:1000), anti-cleaved-caspase3 (ab32042, 1:500), anti-caspase9 (ab184786, 1:1000), Bax (ab32503, 1:1000), Bcl-2 (ab182858, 1:2000), anti-GAPDH (ab8245, 1:2000) and Anti-beta Tubulin (ab6046, 1:5000) were added for overnight incubation. Every antibody utilized was sourced from Abcam in the United Kingdom. Subsequently, the membrane received three washes in 1 × TBST for 5 min each at RT, followed by exposure to secondary antibodies such as HRP-conjugated goat anti-rabbit IgG (ab6721, 1:2000 dilution) or goat anti-mouse IgG (ab6728, 1:2000 dilution) for 1 h at RT. Finally, the application of the ECL reagent (1705062, Bio-Rad, USA) led to the exposure of the membrane, which was subsequently visualized with the Image Quant LAS 4000C gel imaging system (GE, USA). Protein protein levels were assessed by examining the grayscale ratio of the target bands to the reference bands using GAPDH as an internal control with ImageJ software (V1.8.0.112) (Shin et al. 2014). Every test was reiterated on three occasions.
Transcriptome sequencingTPC-1 cells treated with 50 μM Ginsenosides-Rh2 (n = 3) and untreated TPC-1 cells (n = 3) yielded total RNA isolated through Trizol extraction method (Thermo, 16096020, USA). Evaluation of RNA quantity, purity, and intactness was conducted utilizing the Qubit®2.0 Fluorometer® (Thermo, Q33238, USA) with the Qubit® RNA Assay Kit (Shanghai BioGerm, HKR2106-01, Shanghai, China), Nanodrop Spectrophotometer (IMPLEN, USA), and the RNA Nano 6000 Assay Kit with the Bioanalyzer 2100 System (Agilent, 5067–1511, USA), guaranteeing RNA quantity exceeding 100 ng/μL and an A260/A280 ratio within the range from 1.8 to 2.1. Each sample underwent RNA library preparation with an initial total RNA amount of 3 μg. Constructing the cDNA library involved the utilization of the NEBNext® Ultra™ RNA Library Preparation Kit for Illumina® (NEB, E7435L, Beijing, China) and sequencing in accordance with established procedures (Arunachalam et al. 2022; Linkner et al. 2021).
Differential gene expression analysisAnalysis of variance in gene expression was executed based on the transcriptome sequencing data, utilizing the "limma" package in R, applying thresholds of |log2FC|> 1 and P < 0.05. Subsequently, volcano plots displaying differential gene expression were produced with the R package "ggplot2" (Ritchie et al. 2015; Gao et al. 2023).
Gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) enrichment analysisThrough the utilization of R-based tools, the differentially expressed genes (DEGs) underwent enrichment analysis in terms of GO and KEGG pathways. Specifically, the "clusterProfiler" package, "org.Hs.eg.db" package, "enrichplot" package, and "ggplot2" package were employed for this analysis. Visualization of the enrichment outcomes for the three GO classifications, namely Biological Process (BP), Cellular Component (CC), and Molecular Function (MF), was carried out. Additionally, a bubble chart depicting the KEGG enrichment investigation was included (Dai et al. 2022).
Molecular docking simulationMixing 0.1 g of Cu2O powder (566284, Sigma-Aldrich, USA) with 1.5 mL deionized water and 0.2 g polyvinylpyrrolidone (PVP10, Merck, Germany) resulted in the solution. The sample was placed in an ice bath and subjected to sonication with an ultrasonic processor (VCX500, Sonics, USA). Then, 0.2 g of Ginsenosides-Rh2 (Naturex, France) was added to the Cu2O nanoparticle suspension. Sonication was continued under the same conditions (40 kHz, 30 min, in an ice bath) to facilitate Ginsenosides loading onto the Cu2O nanoparticles. A high-speed centrifuge (Eppendorf Centrifuge 5425 R from Germany) was utilized to centrifuge the blend at 110,000 g for a period of 15 min. After discarding the supernatant, the nanoparticles underwent three washes with anhydrous alcohol, with a centrifugation step at 110,000 g for 5 min after each wash to eliminate any remaining solvents. For the final wash, deionized water was used to ensure complete removal of organic solvents. The washed Cu2O@G-Rh2 nanoparticles were then vacuum-dried to complete dryness.
FCM-based analysis of cell cycle characteristicsThe cells that underwent transfection with trypsin and were centrifuged at 300 g for a duration of 5 min were immobilized in 75% ethanol at −20 °C overnight, after two rinses with PBS. Prior to analysis, the cells underwent two rounds of rinsing employing PBS, followed by being re-suspended in 500 μl of 1 × PBS with the addition of 10 μl of 5 mg/ml PI stock solution to reach a final concentration of 50 μg/ml. Subsequent incubation in darkness at RT lasted for 15 min. Employing the BD FACSCanto II flow cytometer (BD Biosciences, USA), the evaluation of cell cycle was accomplished and the data were interpreted with FlowJo software (Zhang et al. 2018).
Construction of Cu2O@Ginsenosides-Rh2 (Cu2O@G-Rh2)The combination of 1 g Cu2O powder (566284, Sigma-Aldrich, USA) with 1.5 mL deionized water solution including 0.2 g polyvinylpyrrolidone (PVP10, Merck, Germany) was enacted. The sample was placed in an ice bath and subjected to ultrasonication using an ultrasonic processor (VCX500, Sonics, USA). Then, 0.2 g of Ginsenosides-Rh2 (Naturex, France) was added to the Cu2O nanoparticle dispersion. The mixture was further sonicated under the same conditions (40 kHz, 30 min, in an ice bath) to facilitate the loading of Ginsenosides onto the surface of Cu2O nanoparticles. Centrifugation of the mixture at 110,000 g for 15 min was performed with a high-speed centrifuge (Centrifuge 5425 R, Eppendorf, Germany). The supernatant was decanted, and the nanoparticles were subjected to a triple wash with anhydrous ethanol three times, each time centrifuging at 110,000 g for 5 min to remove residual solvents. For the final wash, deionized water was used to ensure complete removal of any organic solvent residues. The washed Cu2O@G-Rh2 nanoparticles were then vacuum-dried until completely dry.
At 490 nm, the absorption intensity was gauged with a UV spectrophotometer, and the computation of drug loading content (DLC) and drug loading efficiency (DLE) was carried out applying a previously established Ginsenosides standard curve: DLC (wt%) = (weight of loaded drug / weight of loaded nanoparticles) × 100%; DLE (wt%) = (weight of loaded drug / initial drug input weight) × 100% (Zheng et al. 2021).
Transmission electron microscopy (TEM) for the morphology of nanoparticlesCu2O and Cu2O@G-Rh2 nanoparticles were separately suspended in distilled water and adjusted to a concentration of 0.1 wt%. The suspension was subjected to 10 min of ultrasonication using a VCX500 ultrasonic processor (Sonics, USA) operating at a frequency of 40 kHz to ensure the uniform dispersion of nanoparticles. Subsequently, 10 µL of the nanoparticle suspension was accurately pipetted using a micropipette (4640000, Thermo Scientific, USA) and drop-casted onto clean and dried copper grids (T100C/N, XXBR, China). The samples underwent natural drying at RT for an estimated 5 min to promote the uniform attachment of nanoparticles to the surface of the copper grids.
The samples were subsequently inspected by a TEM (JEM-200CX, JEOL, Japan) operating at an acceleration voltage of 200 kV. To obtain representative data, at least five different regions of each sample were examined to record the morphology, size, and distribution of nanoparticles. The size distribution of nanoparticles was analyzed and documented, confirming the size range of Cu2O@G-Rh2 nanoparticles as 20–30 nm and validating the uniformity and consistency of their morphology (Zheng et al. 2021).
Nanoparticle potentials determinationUtilizing a Zetasizer Nano ZS instrument (Malvern Instruments, UK), zeta potential measurements were executed at 25 °C as the measurement temperature. Each sample was measured three times independently, with 1-min intervals between each measurement to ensure repeatability and accuracy. The Zeta potential values from each measurement were recorded, and calculations of average and deviation were performed to assess the stability and surface charge uniformity of the nanoparticle suspension (Vogel et al. 2017).
X-ray diffraction analysisCu2O@G-Rh2 nanoparticle samples were uniformly spread on an XRD sample holder, ensuring flatness and even distribution to avoid non-uniformity affecting the diffraction data. A copper target (Cu Kα) was used as the X-ray source with a wavelength setting of 1.5406 Å and a scanning range adjusted from 2θ = 20° to 80° to cover the main diffraction peaks of Cu2O and any potentially related to Ginsenosides. Appropriate step size and scanning speed were set to obtain high-resolution diffraction patterns. XRD scanning was performed, and diffraction data were collected. To ensure repeatability and accuracy of the results, each sample underwent at least three independent sample preparations and XRD analyses. The XRD patterns were analyzed to identify characteristic diffraction peaks of the Cu2O crystal structure and any additional diffraction features introduced by the Ginsenosides loading (Nzilu et al. 2023).
UV–visible absorption spectroscopy analysisMeasurements were conducted using a UV–visible spectrophotometer (PS-200, Bolun Jingwei, China). The concentration of Cu2O@G-Rh2 nanoparticles was diluted to 0.05 wt% to prevent oversaturation during UV–visible absorption measurements. The measurement wavelength range was set from 200 to 800 nm, and absorption spectra were analyzed using UVProbe (Shimadzu) to identify characteristic absorption peaks and calculate their intensities and positions (Nzilu et al. 2023).
In vitro release assay of ginsenoside-Rh2The release behavior of ginsenosides from Cu2O@G-Rh2 nanocarriers was determined by dialysis. Initially, 2 mg of ginsenosides containing Cu2O@G-Rh2 was completely dispersed in the release medium (5 mL PBS, pH 5.0) within a dialysis tubing (88242, 3.5 K MWCO, Thermo Scientific, USA). Subsequently, it was placed in a dissolution apparatus and immersed in a release medium consisting of 40 mL PBS (pH 7.0) with 0.5% Tween 80 (w/v), and stirred slowly at 150 rpm at 37 °C. Fresh PBS replacements were collected at 1, 2, 4, 8, 12, 24, and 36 h intervals. The external PBS was measured using the BioTek ELx800 automated enzyme immunoassay analyzer (ELx800, BioTek, USA) to determine the concentration of new ginsenosides based on a calibration curve (Bai et al. 2022).
Detection of H2S contentAfter two washes with PBS, cells were lysed using the H2S Colorimetric Assay Kit (EEA011, Thermo Scientific, USA) to release H2S, and the H2S content within the cells was detected at 665 nm through UV spectroscopy (Ruan et al. 2024).
Detection of GSH contentThe Thermo Scientific GSH Fluorescent Detection assay kit (EIAGSHF, Thermo Scientific, USA) was used to measure GSH content in cells. Incubation of samples or standard solutions with the detection reagent at RT for 15 min resulted in the desired reaction. Measuring the fluorescence outcome involved utilizing a fluorescence microplate reader at 390 nm excitation and 510 nm emission wavelengths. The evaluation began with measuring unbound glutathione GSH, after which a blend was added to change all the oxidized glutathione GSSG to free GSH. The signal generated in response to an excess of the detection reagent accounted for the total GSH content in the sample. The overall GSH level in the specimen was determined according to the signal produced (Ruan et al. 2024).
•OH Fluorescence imagingThe level of hydroxyl radicals (•OH) was detected using the hydroxyphenyl fluorescein (HPF) probe (A4108, Sigma-Aldrich, USA) assay kit. The cell culture received an introduction of 15 μL of a 10 μM HPF probe solution, which was then gently mixed to achieve consistent dispersal. Following that, the cell cultures were maintained at 37 °C with 5% CO2 lasting 45 min. Post-incubation, PBS was used to gently cleanse the cells and eliminate any unattached probes. Imaging was conducted using laser scanning confocal microscopy (CLSM, LSM 510 META, Carl Zeiss AG, Germany) with an excitation range of 490–500 nm and emission range of 515–530 nm in the near-blue and near-green regions, respectively (Ruan et al. 2024). Image analysis was performed using Image J.
LOPs fluorescence imagingLipid peroxides (LOPs) were visualized using the fluorescent probe BODIPY 581/591 C11 (Thermo Scientific, USA) at a dosage of 10 µM, which was introduced to the target cells and then incubated for 30 min to 1 h. Subsequently, PBS rinse (E607008, Sangon, China) was applied to the cells to wash away any unbound probe, following which they were viewed through a fluorescence microscope equipped with 581 nm excitation and 591 nm emission. The obtained images were analyzed using Image J software to assess the levels of lipid peroxides in cells subjected to different treatments. LOPs can induce cell membrane rupture and increase membrane permeability, leading to cell death and thereby inhibiting the survival and proliferation of cancer cells (Ruan et al. 2024).
CRT stainingThe cell fixation procedure involved the use of 4% PFA (E672002, Sangon, China) for a period lasting 15–20 min, succeeded by a 10-min treatment with 0.1% Triton X-100 (A110694, Sangon, China) to facilitate cell membrane permeability. Subsequently, blocking of cells was achieved by incubating them in PBS buffer with the addition of 1% BSA for around 60 min to avoid non-specific binding of antibodies. Subsequent to this, the cells underwent an overnight incubation at 4 °C employing a 1:200 dilution of anti-CRT antibody (C7492, Sigma-Aldrich, USA). After incubation, a fluorescently labeled secondary antibody (Sigma-Aldrich, USA) was applied for 1 h. The fluorescence signal of CRT was detected and recorded utilizing a fluorescence microscope, and the visuals were scrutinized through Image J software (Ruan et al. 2024).
Construction of human immune system-reconstituted miceHIS (Human immune system) mice were developed by transplanting hCD34 + hematopoietic stem cells (hCD34 + HSC) into female NSG (NOD scid gamma) mice aged 4 weeks (Catalog No.: NM-NSG-001, Shanghai Model Organisms Co., Ltd.) following myeloablation. Initially, γ-irradiation (150–170 Gy) resulted in bone marrow suppression. Subsequently, human umbilical cord blood (hUCB)-derived hCD34 + HSCs were intravenously infused into myeloablated mice within one day of isolating the cells from hUCB through Ficoll-Paque Plus density gradient centrifugation for mononuclear cell (MNC) isolation, and the separation of hCD34 + cells from MNC was extended through the application of the human CD34 MicroBead Kit (Miltenyi Biotec, Spain) for magnetic cell sorting. FCM was utilized to assess the purity of hCD34 + cells, employing a CytoFLEX flow cytometer. An equivalent quantity of viable human CD34 + cells were administered into the mice. The successful generation of humanized mice was confirmed when the ratio of hCD45 + cells in mouse PBMC reached 25% or higher (Zhai et al. 2023).
THCA mouse modelsIn this study, humanized mice were utilized to establish a THCA model. Specifically, the right forelimb axillary site of the mice received an injection of 1 × 107 TPC-1 cells. Subsequently, the mice were intravenously administered 100 μg of G-Rh2 and Cu2O@G-Rh2 weekly, beginning seven days post-inoculation. The progression of the tumor was tracked on a weekly basis through the measurement of the minor (a) and major (b) axes applying a caliper. Calculations of tumor dimensions employed the formula π(a2b)/6. After 28 days, the mice were euthanized, and tumor mass was measured using a balance, with three measurements taken per group. Half of each tumor was stored in liquid nitrogen, while the other half was processed for pathological examination. To induce bilateral thyroid tumors, TPC-1 cells were implanted into the right forelimb axillary region to establish primary tumors, followed by injection of the same cells into the left forelimb axillary region five days later to simulate distal tumors (representative of recurrence/metastasis).
Investigating biochemical parameter patternsThe levels of metabolic indicators in mouse serum were determined by means of ALT activity assay kit (E1010, Sigma-Aldrich, USA), AST activity assay kit (E1020, Sigma-Aldrich, USA), BUN assay kit (60–1100, BioAssay Systems, USA), and creatinine assay kit (CR200, Randox Laboratories Ltd, UK). Experimental protocols were implemented in accordance with the instructions given by the manufacturer.
ELISALevels of IL-10, VEGF, IL-6, IL-2, INFγ, TNF-α, perforin, and GzmB in mouse serum were quantified using Mouse IL-10 ELISA Kit (M1000B, R&D Systems, USA), Mouse VEGF ELISA Kit (MMV00, R&D Systems, USA), Mouse IL-6 ELISA Kit (M6000B, R&D Systems, USA), Mouse IL-2 ELISA Kit (M2000B, R&D Systems, USA), Mouse INFγ ELISA Kit (MIF00, R&D Systems, USA), Mouse TNF-α ELISA Kit (MTA00B, R&D Systems, USA), Mouse Perforin ELISA Kit (MBS2506123, MyBioSource, USA), and Mouse Granzyme B ELISA Kit (MBS2023409, MyBioSource, USA). The specific procedures followed the respective kit protocols.
Multicolor FCMThe immunocellular composition of primary and distal tumors was analyzed using multicolor FCM. Tumor samples from each group of mice were digested in HBSS solution comprising 0.5 mg/mL IV collagenase and 0.25 mg/L DNase I for 30 min at 37 °C. Filtering of the digested samples was done via a 40 μm cell strainer, subsequent to which centrifugation at 400 g was carried out for 10 min. Cells were then blocked with Fc blocking antibodies (BioLegend) for 15 min to prevent nonspecific binding and subsequently stained with fluorescence-labeled monoclonal antibodies. Antibodies used comprised FOXP3 Alexa Fluor® 488, CD4 PE-Cy5, CD25 PE, CD45 PerCP/Cy5.5, CD8 Brilliant Violet 650, CD3 PE/Cy7, CD86 FITC, and CD80 APC purchased from BioLegend. After staining, samples underwent cleansing utilizing PBS enriched with 1% BSA and were redispersed in 500 μL PBS. The FCM assay was executed utilizing the BD FACSAria Fusion Flow Cytometry Cell Sorter (BD Biosciences), and FlowJo LLC provided FlowJo v.10 software for the data analysis process (Zhai et al. 2023).
Histological analysis H&E stainingSpecimens of tumors obtained from every mouse group underwent fixation in 10% neutral formalin, paraffin embedding, and subsequent sectioning after deparaffinization with xylene. Subsequently, the sections were treated with H&E for staining, dehydrated in 95% ethanol, counterstained with eosin, dehydrated in a graded series of ethanol, cleared in xylene, air-dried, embedded in neutral resin, and scrutinized under an optical microscope (Wang et al. 2021).
ImmunohistochemistryRegular immunohistochemical staining methods were applied to tumor specimens from every mouse cluster (Xie et al. 2022) using antibodies specific for Ki-67 (1:200, ab15580, Abcam, UK), CHEK1 (1:200, ab40866, Abcam, UK), GPX4 (ab125066, 1:200, Abcam, UK), DLAT (PA5-112,114, 1:100, Thermo Fisher Scientific, USA), and FDX1 (PA5-59,653, 1:150, Thermo Fisher Scientific, USA). The software ImageJ was employed for the analysis of images to determine the portion of Ki67 + cells identified as positive and to gauge the levels of alternative proteins based on grayscale intensity.
Tunel assayEach mice category's tumor tissue sections were processed for TUNEL staining with a one-step TUNEL apoptosis detection kit (C1086, Beyotime, Shanghai). Upon dehydration completion, 4% PFA was employed for fixing the slides at RT over a 30-min interval. Subsequent steps involved washing with PBS, introduction of permeability using 1% Triton X-100 at 4 °C for 4 min, and labeling with terminal deoxynucleotidyl transferase (TdT) accompanied by labeled nucleotides at 37 °C in darkness for 60 min. Upon completion of the PBS wash, the sections were counterstained with DAPI for 5 min. TUNEL-positive cells appeared as green fluorescence under a fluorescent microscope (Zhou et al. 2022). Image analysis using ImageJ software was performed to calculate the TUNEL-positive cell rate as TUNEL-positive cell rate (%) = (number of TUNEL-positive cells in total cells) × 100%.
Immunofluorescence StainingSections of tumor tissue in mice groups were treated with Alexa Fluor® 647-conjugated Anti-CD8 antibody (ab237365, 1:100, Abcam, UK) at 4 °C for an overnight incubation. Subsequently, washing with PBS was conducted on the sections, which were then incubated in darkness for 2 h using the secondary antibody goat anti-rabbit IgG H&L (HRP) (ab6721, 1:1000, Abcam, UK). Subsequent removal of surplus antibody through washing was followed by counterstaining with DAPI (1:1000, #8961, Cell Signaling) in darkness for an hour. Subsequent to cleansing with PBS, the slices were observed under a confocal microscope (Leica, STELLARIS 5, Germany) (Hossain et al. 2015). Image analysis using ImageJ software was performed to calculate the fluorescence intensity of CD8.
Statistical analysisOur research harnessed R coding language version 4.2.1 executed through the RStudio development suite version 2022.12.0–353. Utilizing GraphPad Prism 8.0, the data underwent analysis and the results are showcased in the form of mean with standard deviation (Mean with SD). The analysis involved using unpaired t-tests to compare pairs and one-way ANOVA to compare multiple groups. Homogeneity of variance was examined through Levene's test, with Dunnett's T3 and LSD-t tests applied for post hoc pairwise comparisons under homogeneity of variances and Dunnett's T3 test under heterogeneity of variances. Statistically noteworthy differences between groups were identified at a significance level of P < 0.05 (Zhang et al. 2020).
留言 (0)