
記住我
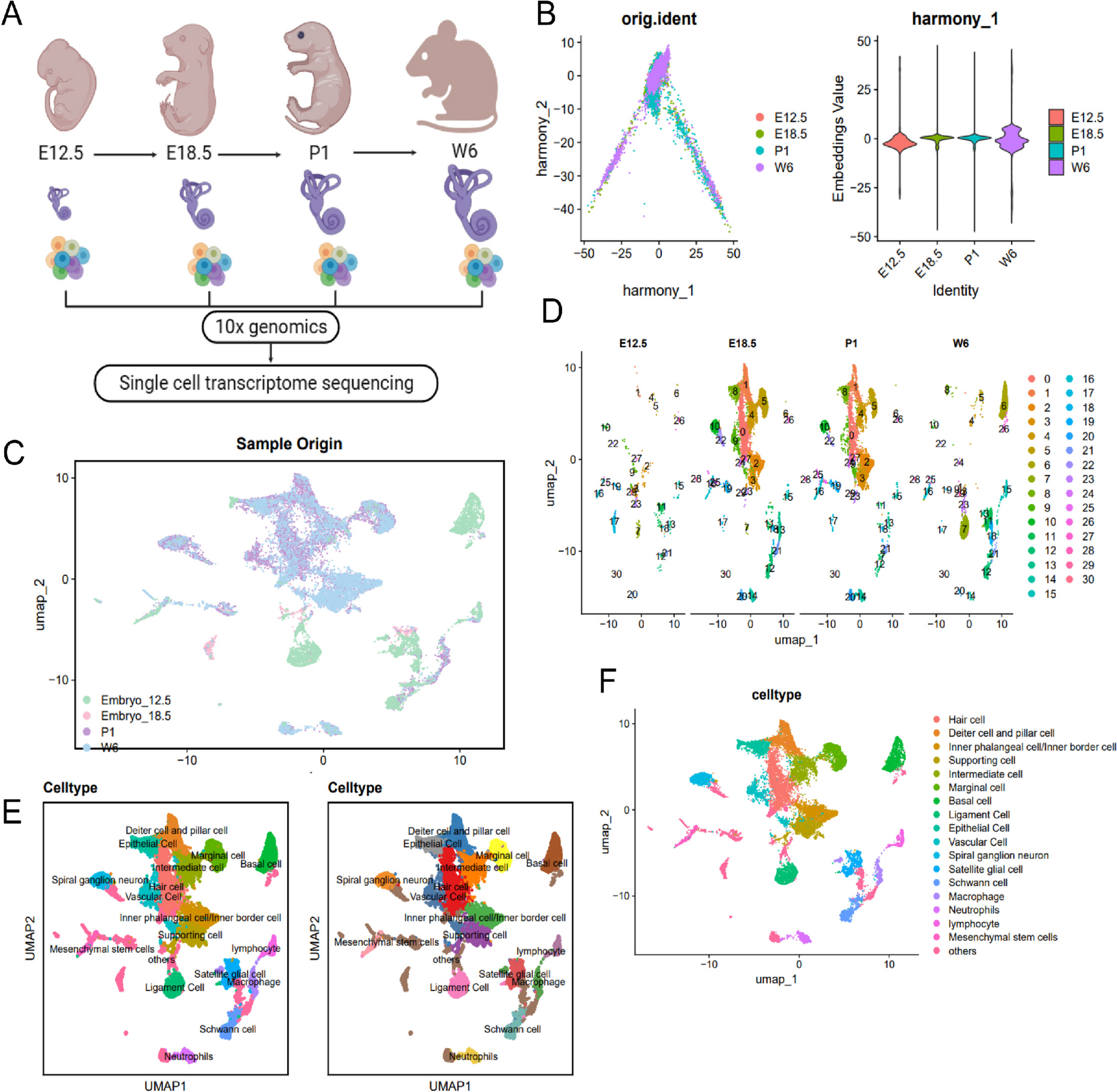






Sourced from Beijing's Vital River Laboratory Animal Technology Co., Ltd. (Strain code: 219), female and male C57BL/6 mice, weighing 23 ± 2 g and aged between 6 and 8 weeks, were housed in the same cage for mating facilitation. Maintaining specific pathogen-free (SPF) conditions, all mice resided in an animal facility where the humidity ranged from 60 to 65% and the temperature was between 22 and 25℃. Unrestricted food and water were available to the mice throughout their housing. The experiments commenced after the mice had acclimated for a week, based on an evaluation of their health conditions. The Animal Ethics Committee of Shengjing Hospital at China Medical University approved the experimental procedures.
The experiment consisted of four groups (n = 1), namely, the E12.5 group (embryos developed to the stage of E12.5), the E18.5 group (embryos developed to the stage of E18.5), the P1 group (first day after birth), and the W6 group (6 weeks of age). Euthanasia of mice in each group took place post anesthesia, with the cochlear tissues being harvested for subsequent research endeavors (Petitpré et al. 2022).
scRNA-seq and data analysisSurgical blades were utilized to fragment cochlear tissues extracted from individual mouse groups. Single-cell suspensions were generated from the tissues by employing trypsin (Sigma-Aldrich, 9002–07-7). The C1 Single-Cell Auto Prep System (Fluidigm, Inc., South San Francisco, CA, USA) was utilized to isolate individual cells. Lysis of the cells took place within the chip upon capture, leading to the liberation of mRNA, which was then transformed into cDNA by reverse transcription. Post lysis and reverse transcription, the cDNA underwent preliminary amplification using microfluidic chips with the aim of sequencing. Library construction and single-cell sequencing on the HiSeq 4000 Illumina platform were performed applying the pre-amplified cDNA, incorporating paired-end reads of 2 × 75 bp in length and an approximate read count of 20,000 reads for each cell (Mahmoudi et al. 2019).
The "Seurat" package within the R software framework was harnessed for data analysis procedures. Quality control measures were applied to the data based on the standards of 200 < nFeature_RNA < 5000 percent.mt < 20. This was followed by the identification of 2000 genes that exhibited significant variability in their expression variances (Yang et al. 2023).
The reduction in dimensionality of the scRNA-seq dataset was achieved through the utilization of principal component analysis (PCA) on the expression data of the top 2000 genes showing high variability. Utilizing the Elbowplot function within the Seurat software package, we opted for the extraction of the foremost 20 principal components (PCs) to facilitate subsequent analyses. Major cell subgroups were identified using the FindClusters function within the Seurat package at the default resolution of 1. The scRNA-seq data underwent non-linear dimensionality reduction via the application of the t-distributed Stochastic Neighbor Embedding (t-SNE) algorithm in the next stage. The Seurat tool was instrumental in pinpointing marker genes associated with distinct cell subgroups, and the "SingleR" package was employed to annotate these clusters of cells based on the marker genes (Ma et al. 2019). Additionally, pseudotime analysis was implemented using the monocle2 platform.
Cell cultivationBLUEFBIO provided the HEI-OC1 and Ana-1 cells. The growth environment for HEI-OC1 cells involved incubation at 37 °C in a humidified incubator with 5% CO2, utilizing high-glucose Dulbecco's Modified Eagle Medium (11,965,118, Gibco) free of antibiotics and supplemented with 10% fetal bovine serum (FBS, 10,270,106, Gibco). Passage of cells took place with the application of 0.25% trypsin/EDTA (T4049, Sigma-Aldrich) upon reaching 80% confluency. The initiation of cellular senescence was prompted in HEI-OC1 cell populations through exposure to 1 mM H2O2 from Sigma-Aldrich (386,790-M) over a 1-h period within 48-well cell culture dishes (Lin et al. 2019, Cho et al. 2022).
Utilizing the RPMI 1640 medium (11,875,093, Gibco), Ana-1 cells were subjected to cultivation supplemented with 10% FBS (10,270,106, Gibco) and 1% penicillin–streptomycin (15,070,063, Gibco). The 37 °C incubation of cells occurred in an environment with 5% CO2 and humidity. To induce inflammation, the activation of Ana-1 cells was initiated by exposure to lipopolysaccharide (LPS, HY-D1056, MCE) at a concentration of 1 μg/ml for a duration of 24 h. Ana-1 cells were subjected to fluorescence labeling as per the manufacturer's guidelines, employing CellTracker™ Deep Red Dye (C34565, Invitrogen). Briefly, the treatment involved the incubation of 1.5 × 105 Ana-1 cells in a medium devoid of serum, followed by the addition of a dye solution at a concentration of 500 nM. Post a 30-min incubation, the cells received a PBS wash and were subsequently placed in 8-well chamber slides for seeding. Representative images were captured using fluorescence microscopy at 0 and 24 h after seeding CFSE-EVs (5 × 104 EVs per seeded cell) (Ragni et al. 2020).
For the isolation and culture of MSC, the leg bones of 4-week-old female C57BL/6 mice, weighing approximately 23 ± 2 g, were flushed to extract bone marrow cells. The isolated cells were then resuspended, and red blood cell lysis buffer (00–4333-57, Invitrogen) was utilized to eliminate red blood cells from the sample. The culture of about 5 × 106 cells was undertaken in α-Minimum Essential Medium (MEM α) with the addition of 9% horse serum (16,050,130, Gibco), 9% fetal bovine serum (10,270,106, Gibco), 1% Gluta-Max (35,050,079, Gibco), and 1% penicillin–streptomycin (15,070,063, Gibco). Culture conditions included maintaining the cells at 37 °C in a humidified CO2 incubator. MSCs attached to the culture dishes were collected. A digestion process utilizing 0.25% trypsin/EDTA (T4049, Sigma-Aldrich) was executed on the cells, after which they were cultured and propagated for three rounds, with the medium renewal taking place every 3–4 days. Referred to as MSCs were these specific cells. To induce differentiation of MSCs into auditory cells, the cells were cultured with 20 ng/ml EGF (HY-P7067, MCE) and 50 ng/ml IGF-1 (HY-P70698, MCE) for 2 weeks. This was succeeded by the introduction of 10 ng/ml bFGF (HY-P7066, MCE) for an additional 2 weeks to facilitate the differentiation of MSCs (Jeon et al. 2007). To generate GFP-tagged MSC, the lentiviral expression vector pLV/puro-EF1a-GFP was introduced into MSCs through transfection facilitated by X-treme GENE HP reagent (6,366,244,001, Sigma-Aldrich) under the recommended procedures. GFP-tagged MSCs underwent flow cytometry-based sorting after the completion of a 3-day transfection (Wang et al. 2018).
Cell grouping and transfectionTo establish cell lines with gene silencing or overexpression, we employed lentiviral transduction. These included MSCs with knockdown of Apelin (sh-Apelin) and their corresponding control cell line (sh-NC), as well as MSCs with overexpression of Apelin (oe-Apelin) and their control cell line (oe-NC). Two sets of shRNA sequences were concurrently tested for a knockdown, and the more effective set was chosen for subsequent experiments. Table 1 illustrates the lentiviral sequences implemented for the silencing of genes. Shanghai Bioscientific (China) offered plasmids and lentiviral packaging solutions.
Table 1 Lentiviral transfection sequenceThe initiation of lentivirus-mediated cell transfection involved the seeding of 1 × 105 cells into a 6-well plate. The transfection process commenced when the cellular density reached 70–90%, with the introduction of a medium containing an adequate dose of enclosed lentivirus (MOI = 10, operational concentration around 1 × 106 TU/mL) and 5 μg/mL polybrene (TR-1003, Sigma-Aldrich). An equivalent medium volume was incorporated after 4 h to alleviate the polybrene concentration, and a fresh medium exchange occurred after 24 h. Stable transduced cell lines were established through resistance screening with 1 μg/mL puromycin (A1113803, Thermo Fisher) following a 48-h transfection (Zhi et al. 2021, Gao et al. 2022).
β-galactosidase stainingTo determine cellular senescence, the Cell Senescence β-Galactosidase Staining Kit (C0602, Beyotime) was utilized as directed by the manufacturer for assessment. The seeding of cells took place in a 6-well culture dish. Following a gentle wash with PBS, each well was treated with 1 mL of fixation solution for β-galactosidase staining, and then fixed at room temperature for 15 min. Subsequent to the triple PBS washes, 1 mL of the staining working solution was administered to every well containing the cell samples. Overnight incubation at 37 °C without CO2 was conducted in the 6-well plate, which had been meticulously covered with foil. Microscopic observation was performed, and 5 fields of view in the center and periphery were counted and averaged (Cho et al. 2022).
CCK-8 assayThe CCK-8 assay kit (CK04, Dojindo Laboratories) was applied for the analysis of cell viability. The seeding density of cells in a 96-well plate was set at 1 × 105 cells per well. Each well received 10 μL of CCK-8 solution daily, with subsequent addition of 100 μL of serum-free medium. An incubation period of 2 h at a temperature of 37 °C was allowed for the plate, followed by measuring the optical density (OD) at a wavelength of 450 nm. Viability of cells was assessed by the formula % = [(experimental well—blank well)/(control well—blank well)] × 100% (Ma et al. 2019).
ELISA assayThe ELISA kits were employed to measure the concentrations of IL-10 (IL-10, CUSABIO), Arg1 (CSB-EL002005MO, CUSABIO), IL-1β (CSB-E08054m, CUSABIO), and IL-6 (ab222503, Abcam) in the cell culture supernatant. First, the antigens were diluted to appropriate concentrations and added to the wells of an enzyme-labeled reaction plate. Then, the enzyme-labeled antibodies and substrate solution were applied to the plate, incubated, and followed by the introduction of 50 μL stop solution to halt the reaction, with the experimental outcomes assessed in a span of 20 min. The ELISA reader (1,681,135, Bio-Rad, USA) was utilized for reading the plate's absorbance at 450 nm. After plotting a standard curve, thorough analysis was conducted on the obtained data (Yun et al. 2021). All assays were conducted following the manufacturer's guidelines. Each sample was tested in triplicate to ensure result accuracy.
Acridine orange (AO) stainingCell viability was determined using AO staining solution (CA1143, Soleibao). The experimental procedure was conducted according to the instructions provided by the reagent. Initially, two PBS washes were administered to the gathered cells. Subsequently, the number of cells was tallied and modified to a density of 1 × 106/ml. The infusion of AO dye (1 mg/ml) into the cell suspension led to a concentration of 10 µg/ml. Post a 15-min incubation under light exclusion at room temperature, the stained cells underwent observation under a fluorescence microscope (with an excitation filter set at 488 nm and an emission filter at 515 nm) in order to compute cellular vitality. Apoptotic cells appeared orange, while viable cells appeared green. Five fields of view were observed in both the center and periphery and the average values were obtained (Niknazar et al. 2019). Each experiment was replicated thrice.
Collection, preparation, and depiction of EVsMSC groupings received two PBS washes and were subsequently exposed to FBS-free MDEM after incubation. The supernatant was gathered after a span of 48 h, then centrifuged to separate cells and debris. At a temperature of 4 °C, the centrifugation process involved steps at 300 g for 10 min, 2000 g for 20 min, and 10,000 g for 30 min. A 1-h exposure at 37 °C was employed for the labeling of cells with 10 μM CFSE (65–0850-84, Invitrogen). Ultracentrifugation at 4 °C and 100,000 g for a period of 3 h was performed to isolate CFSE-labeled EVs. Following the removal of any extra dye, the EVs that had been collected underwent a washing process with PBS and were then reconstituted in PBS. To ensure the purity of the isolated EVs sample, we evaluated the ratio between the number of microvesicles and the total amount of protein. Only when the ratio fell within the range of 108–1010 particles/µg protein did we consider the EVs batch to have high purity (Ragni et al. 2020, Liu et al. 2020).
Fluorescently labeled EVs (CFSE-EVs) underwent examination through a CytoFLEX flow cytometer (Beckman Coulter, USA) prepared with FITC-fluorescent beads for calibration. This enabled the identification of fluorescent particles with a size as minute as 100 nM. Standard beads of 160, 200, 240, and 500 nM (Biocytex) were used for calibration. EVs were diluted in PBS at a 1:10,000 ratio and stained with CD63-APC (143,905, Biolegend) and CD81-APC (104,909, Biolegend) antibodies. A 30-min staining procedure was conducted at a temperature of 4 °C. Post-staining, the samples underwent dilution in PBS to 1,000 μL for assessment with the flow cytometer.
To establish a gating strategy for selecting only the stained EVs, CFSE-EVs were compared to PBS + CFSE samples in the FITC channel. Events within this gate were used for subsequent analysis. The purpose of this step was to exclude antibody aggregates that produced spontaneous fluorescence in the FITC channel, ensuring that only CFSE-labeled EVs were analyzed. Subsequently, CFSE-positive EVs were detected using the flow cytometer (Ragni et al. 2020).
Nanoparticle tracking analysis (NTA)Dilution in PBS at a 1:100 ratio was applied to the EV samples. The diluted EV samples were then placed in the Nanosight LM10-HS system (Malvern Panalytical). For each EV sample, a 30-s video recording was conducted to capture the movement trajectory of EV particles in PBS (Ragni et al. 2020). The experiment was repeated three times.
Atomic force microscopy (AFM)Firstly, the collected exosomes were diluted in PBS and then dropped onto pre-prepared glass slides. Subsequently, the samples were air-dried overnight at room temperature. Next, high-resolution images of the exosomes were prepared using an AFM and an ACT cantilever in semi-contact mode (Bagheri et al. 2020).
Transmission electron microscopy (TEM)10 μL of EV suspension was fixed in 4% paraformaldehyde and drop-casted onto formvar carbon-coated EM grids for air-drying. Subsequently, it was stained with 3% phosphotungstic acid (G1871, Soleibao) for 5 min. Finally, the morphology of EVs was thoroughly observed and analyzed using a JEOL 1010 TEM (JEOL) (Yang et al. 2023).
Immunofluorescence (IF) stainingThe cells obtained were treated with 4% paraformaldehyde (P0099, Beyotime) for a duration of 15 min and then rinsed thrice in PBS. The cochlear tissues of mice, post-embedding, underwent slicing into slices of 8 µm, cleansing with PBS on slides arranged in advance, and then fixation with 4% paraformaldehyde lasting 10 min per slide to boost the adhesion of the tissue to the glass material. Post three subsequent PBS washes, cells or sections of the cochlea were subjected to hindering by a mixture including 0.3% Triton X (BL935B, Biosharp), 1% bovine serum albumin (V900933, Sigma-Aldrich), and 10% serum from goats (C0265, Beyotime) for an interval of 30 min. Overnight incubation at 4 °C involved treating the slides with the primary antibodies provided: Prestin (PA5-103,158, 1/100, ThermoFisher), Arg-1 (ab137614, 1:100, abcam), Myo7a (ab150386, 1/100, Abcam), Sox2 (ab92494, 1/100, Abcam), Iba1 (ab178846, 1/200, Abcam), Na–K ATP (A11683, 1/200, ABclonal), Atoh1 (PA5-29,392, 1/100, Invitrogen), CD206 (MA5-16,871, 1/100, Invitrogen), CD80 (MA5-15,512, 1/200, Invitrogen), and Cx26 (71–0500, 1/100, Invitrogen). Upon completion of three PBS rinses, the slides were subjected to a 2-h incubation at room temperature in the absence of light, with the addition of secondary antibodies: goat anti-rabbit IgG (H + L) Alexa Fluor® 488 (ab150077, 1/200, Abcam), Alexa Fluor® 594 (ab150080, 1/200, Abcam), or Alexa Fluor® 647 (ab150083, 1/200, Abcam). The staining of F-actin involved the use of Alexa Fluor® 647-conjugated phalloidin (A22287, 1:200, sourced from Invitrogen) and incubated for 30 min at ambient temperature. Upon conclusion of the experimental steps, the slides underwent a thorough wash with PBS before being affixed using an anti-fade mounting compound infused with DAPI (4083, CST). Subsequent observations and image capture were performed utilizing a laser scanning confocal microscope (LSM 980, ZEISS) (Yang et al. 2022, Takeda et al. 2021). Five animals were assigned to each group, and the cochlear tissue of each animal was stained on a singular slice, with an individual field of view designated for imaging. The cellular studies were redone on three occasions. ImageJ software from the National Institutes of Health in the USA was employed to estimate the proportion of positive cell density subsequent to imaging.
Real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR)Extraction of total RNA from cellular samples in the various groups was accomplished through the application of the Trizol reagent (T9424, Sigma-Aldrich). A UV–visible spectrophotometer (ND-1000, Nanodrop, USA) was employed to measure the quality and concentration of RNA. The mRNA expression analysis entailed the implementation of reverse transcription with the PrimeScript™ RT reagent kit (RR014B, TaKaRa, Japan). The performance of RT-qPCR was executed via an ABI 7500 PCR apparatus (Applied Biosystems, USA), with the utilization of the TB Green® Premix Ex TaqTM reagent kit (RR420W, TaKaRa, Japan). The internal control for mRNA was GAPDH. Employing the comparative Ct method for relative quantification, the fold change in target gene expression in the experimental group as opposed to the control group was represented using the formula 2−ΔΔCt, where ΔΔCT = ΔCt experimental group—ΔCt control group, and ΔCt = target gene Ct—internal control gene Ct (Yang et al. 2023).
Western blot (WB)The protein extraction kit (BC3710, Solarbio, China) was applied for the extraction of total cellular or tissue proteins. Centrifugation of the supernatant was carried out at 13,000 rpm for 15 min at a temperature of 4 °C. Utilizing the BCA protein quantitation kit (P0010, Beyotime, China) allowed for the determination of protein levels. A PVDF membrane was used for the transfer of proteins following their separation by SDS-PAGE electrophoresis. Blocking the membrane involved the utilization of 5% skim milk at ambient temperature for a duration of 60 min. Addition involved diluting the primary antibodies listed below: Apelin (PA5-114,860, 1/1000, ThermoFisher), Prestin (PA5-103,158, 1/1000, ThermoFisher), Atoh1 (21,215–1-AP, 1/1000, ThermoFisher), Sox2 (ab92494, 1/1000, Abcam), Myo7a (ab150386, 1/1000, Abcam), p53 (ab26, 1/1000, Abcam), p21 (ab109199, 1/1000, Abcam), Arg-1 (ab203490, Abcam), IL-10 (ab310329, Abcam), and GAPDH (ab8245, 1/5000, Abcam). Anti-Rabbit-IgG secondary antibody (7074, 1/1000, CST) or Anti-Mouse-IgG secondary antibody (7076, 1/1000, CST) was then added and incubated at ambient temperature for 1 h. The membrane underwent three wash cycles applying TBST solution (each lasting for 5 min). Removal of TBST was followed by preparation of an adequate volume of ECL working solution (WBULS0500, EMD Millipore, USA). The PVDF membrane was introduced into the ECL developing solution for soaking and then kept at room temperature for 1 min. Elimination of excess ECL operating solution from the PVDF film, enclosing the film with plastic wrap, placing it in a dark storage, and allowing a 5–10 min exposure for the development and fixing (Wang et al. 2021). Quantitative grayscale analysis of the WB bands was achieved by employing the ImageJ software, with GAPDH serving as the internal reference.
Flow cytometryPreparation for flow cytometry-based cell surface marker analysis involved isolating MSCs with 0.25% trypsin/EDTA (T4049, Sigma-Aldrich) and placing them in PBS supplemented with 3% FBS. Under a cold condition of 4 °C, cells were treated with specific antibodies for staining: anti-CD11b-PE (553,311), anti-CD34-PE (551,387), anti-CD29-PE (562,801), anti-CD45-PE (567,111), anti-CD90-PE (551,401), anti-CD105-PE (562,759), anti-CD146-PE (562,196), anti-Sca-1-PE (561,077), anti-CD80-APC (560,016), and anti-F4/80-APC (566,787) purchased from BD Biosciences. Anti-CD63-APC (17–0631-82), anti-CD81-FITC (MA5-17,939), anti-CD206-APC (17–2061-82), and anti-CD163-PE (12–1631-82) were purchased from Invitrogen. Finally, each group's cells underwent two PBS washes, were sorted via a flow cytometer (Beckman Coulter, USA), and analyzed utilizing the FlowJo software (Liu et al. 2020).
Animal experiments, MSCs transplantation, and EVs treatmentCBA/CaJ (000654) male mice were purchased from Jackson Laboratory, including young (8–10 weeks old) and old (18–19 months old) mice. The entire mouse population was housed in a facility that met SPF standards, where the humidity was controlled within 60–65% and temperatures were maintained between 22–25 °C, with water freely available. A one-week acclimation period was provided for the mice before the experiment, with careful observation of their health status. The institutional committee responsible for animal ethics approved the experimental procedures and protocols for animal usage (Seicol et al. 2022).
For MSCs transplantation: Anesthetized mice were positioned on a warming mat to regulate their body temperature. The skin in the area behind the left ear, where the procedure was performed, was disinfected with 1% iodine solution. The untreated control was implemented on the right ear. By utilizing a Leica M205A stereomicroscope, a slice of 0.5 cm was initiated on the rear aspect of the left ear, and the subcutaneous muscle was dissected to expose the temporal bone. Next, the utilization of forceps allowed for the exposure of the round window vicinity by opening the left ear canal, and the diameter of the opening was expanded to facilitate the visualization of the round window membrane (RWM). The delivery of MSCs suspension into the Scala tympani of the ear through the RWM was accomplished by employing a Picoliter microinjection system (PLI-100A, Warner Instrument) from a glass pipette within a 1 to 2-min timeframe. Approximately 0.5 μL (1 × 107 cells/mL) of cell suspension was injected into the cochlea. Upon withdrawal of the pipette, a tiny fragment of muscular or adipose tissue obtained from the neck region was promptly positioned above the puncture area to prevent any leakage from the circular window site. Closure of the auditory sac orifice involved the application of 6–0 absorbable chromic sutures, after being concealed by adipose tissue. Post the treatment, the animals were reinstated within their enclosures for staying (Takeda et al. 2021).
For EV treatment: Administration of EVs.sh-NC or EVs.sh-Apelin, both at a concentration of 60 μg/mL, via tail vein injections was conducted on a cohort of 5 mice for a continuous duration of 4 weeks (Li et al. 2021).
The assessment of auditory brainstem response (ABR) and distortion product otoacoustic emissions (DPOAE) values was performed at intervals of 7, 14, and 28 days post MSCs engraftment or EV treatment in the respective mouse groups. The mice were euthanized on the 28th day of the trial through cervical dislocation while under general anesthesia, and tissue specimens were gathered for subsequent experiments (Li et al. 2021).
ABRIn this study, administering pentobarbital sodium intraperitoneally at a dose of 50 mg/kg was used to induce anesthesia in mice. The body temperature of 37 °C was sustained through the application of a heating pad. Upon initiating the test, a 1/4-inch microphone (PCB-378C01, PCB Piezotronics) was placed on the ears of the experimental animals. Additionally, three silver wire electrodes were inserted: an active electrode between the ears at the cranial vertex, a reference electrode beneath the right ear, and a ground electrode in the midline of the back. During the experiment, stimuli of different tones including 2, 4, 8, 16, and 32 kHz were delivered at a rate of 21.1 kHz per second. The BioSigRZ software (TDT, USA) was employed for capturing the animals' responses. Starting at 90 decibels sound pressure level (dB SPL) for the initial stimulation intensity, it was subsequently diminished in 5 dB increments until achieving the threshold level. The threshold was designated as the minimum level of sound intensity required for observing visually identifiable and replicable wave patterns (Sun et al. 2022).
DPOAEThis investigation involved the measurement of 2f1-f2 DPOAE responses through the implementation of the real-time signal processing system II developed by Tucker-Davis Technologies (TDT, USA). The output stimulus was calibrated using ABR measurements to ensure accurate measurements. The primary stimulus signals (f1 and f2) were set at two intensity levels, specifically L1 = 80 dB SPL and L2 = 75 dB SPL, with an f2/f1 ratio of 1.22. DPOAE response thresholds were recorded at multiple frequency ranges, including 2, 4, 8, 16, and 32 kHz. The DPOAE recordings were executed by employing two separate speakers to emit the primary tones, along with the utilization of a low-noise microphone, ER 10B (Etymotic Research, USA). In DPOAE analysis, only when the peak of the 2f1-f2 spectral component exceeded the background noise level by more than 3 dB was it considered a valid DPOAE response (Sun et al. 2022).
Methods for analyzing data and statistical softwareOur research involved the application of R language version 4.2.1 via the integrated development interface RStudio, version 2022.12.0–353. The utilization of GraphPad Prism 8.0 was instrumental in processing all data. Demonstrative data was exhibited in the format of Mean ± SD, denoting the mean with standard deviation. The utilization of non-paired t-tests was essential for the evaluation of two datasets, whereas one-way analysis of variance (ANOVA) was pivotal for investigating differences among multiple datasets. Levene's test was utilized to evaluate the equality in variances. Pairwise comparisons underwent Dunnett's T3 and LSD-t tests provided that variances showed homogeneity; however, in the absence of homogeneity in variances, the Dunnett's T3 test was employed. The statistical significance level was established at P < 0.05, revealing meaningful distinctions across the analyzed groups (Zhang et al. 2020).
留言 (0)