
記住我
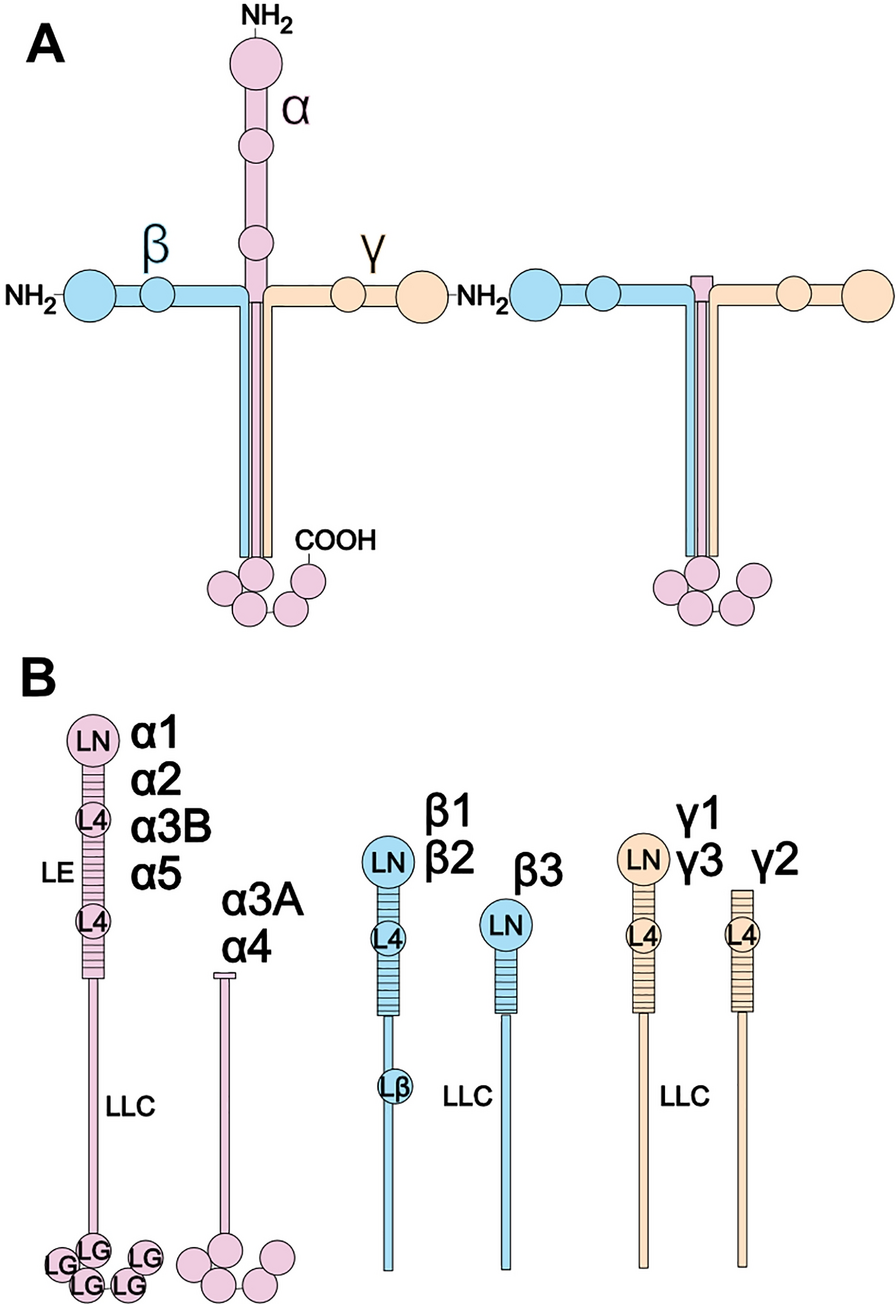






While mutations in genes encoding various cellular components are well recognized as drivers of malignant transformation, mutations in genes encoding ECM proteins are not typically regarded as cancer-causing factors. However, evidence points to a prominent role of LMs in various stages of oncogenesis, including the acquisition of the transformed phenotype. As summarized in Table 1, alterations in the expression or localization of many LMs and specific LM chains have been implicated in the progression of most, if not all, human cancers. In the following sections, we will provide a comprehensive review of these changes in LM expression across the main cancer types, the epigenetic and post-transcriptional regulation of LM chains, and the role of limited proteolysis of LM chains within the tumor interstitial space. Additionally, we will explore the significance of these processes in malignant transformation, metastasis, the acquisition of aggressive cancer hallmarks, and their impact on the prognosis of major human malignancies.
Table 1 Summary of the effects of laminins and their subunits in human cancersBreast cancerBreast cancer is among the most common cancers globally, with a higher prevalence in women. At the molecular level, breast cancer is classified into three subtypes based on hormone receptor presence (hormone receptor positive; HR+), human epidermal growth factor receptor-2 expression (HER2+), or the absence of these markers, known as triple-negative breast cancer (TNBC). While HR+ subtypes often exhibit higher laminin levels, the interaction between laminins and breast cancer is complex and influenced by factors within the TME.
Different LMs have been implicated in the breast carcinogenesis. One of the first studied was LM-111, since it was found that whereas mammary epithelial cells form polarized colonies resembling normal breast acini on 3D LM-111-rich gels, breast cancer cells form disorganized tumor-like structures [42]. Later, it was found that myoepithelial cells from healthy breast tissue promoted luminal breast epithelium polarity and BM deposition more effectively than tumor-derived myoepithelial cells [43]. These differences were associated with reduced LM-111 deposition by tumor-derived compared to normal myoepithelial cells. Importantly, restoration of colony architecture in LM-111 gels by downmodulation of integrins or oncogenic receptors in cancer cells, leads to reversion of the malignant phenotype and the formation of polarized growth-arrested colonies [44]. These findings underscore the central role of LM-111 in myoepithelial-luminal cell interactions and suggest alterations in LM-111 deposition and basement membrane formation may contribute not only to breast cancer progression but even to malignant transformation. Supporting this, loss of cell-surface LM-111 anchoring is linked to increased tumor growth and poor clinical outcomes in breast and brain cancer patients, leading to cancer cell proliferation and chemotherapy resistance [45]. The low cancer cell attachment to LM-111 may be partially related to the dysfunctional glycosylation of α-DG due to acetylglucosaminyl-transferase (LARGE1/2) enzyme downregulation [45], a feature observed in several advanced cancers.
LM-332 has been also implicated in breast cancer progression, though evidence is partially contradictory. Initial immunohistochemical (IHC) studies suggested reduced LM-332 expression in breast adenocarcinomas compared to healthy breast tissue or benign epithelial lesions [46, 47]. From these data, the authors inferred that reduced LM-332 expression might contribute to cancer progression by altering interactions between cancer cells and the surrounding environment. High LM-332 levels can be associated with a better prognosis, especially in luminal breast cancer subtypes. However, later studies indicated upregulation of LM-332 in distinct breast cancer subtypes, particularly in TNBC and HER2+ tumors, and its association with increased motility and invasion through enhanced β1 and β4 integrin signaling in cancer cells [48,49,50]. Supporting a pro-tumor role of LM-332, malignant transformation of breast epithelial cells has been associated with autocrine production of this LM-332, providing a critical survival signal to rescue cancer cells from anoikis through activation of the α6β4 integrin/Rac/NF-κB pathway [51]. Notably, reversion of breast tumor cells to normal phenotype requires the upregulation of HOXD10 and the downregulation of the NF-κB pathway, which antagonistically regulate the expression of miR-34c, miR-30e and miR-144 [52]. In this way, upregulation of these miRs inactivates both EIF5A2 and SCA1-induced transcription of matrix MMP-9, leading to BM stabilization. Regression of TNBC has been also associated with upregulation of miR-17/20a, which targets the LM-α3 chain [53], reinforcing the idea that high LM-332 levels promotes breast cancer progression. Curiously, LM-332 stabilization has also been implicated in the breast cancer cell phenotype reversion [52].
LM-332 upregulation mainly occurs in the stroma, particularly in cancer-associated fibroblasts (CAFs) [54]. This suggests that LM-332 promotion of cancer cell movement and tumor dissemination is primarily due to its upregulation in the TME [26]. LM-332 induces metastasis through various mechanisms, including activation of signaling pathways downstream of the α3β1 integrin, leading to cytoskeletal remodeling involving PI3K and the Rho family of small GTPases [48], increased expression of MMP-2 and MMP-9 [26], and activation of signaling pathways regulating EMT [55].
Although most previous studies did not differentiate between LM-α3 chain isoforms, α3A is the predominant isoform in breast epithelium [26]. Nevertheless, LMs consisting of the α3B chain combined with β1, β2, and γ1, have also been implicated in breast cancer by influencing stromal cell function. Specifically, LM-3B11 exerts a distinct effect on microvascular ECs [56]. Interestingly, recombinant LM-3B11 exhibits relatively weak cell adhesion activity through both α3β1 and α6β1 integrins. Upon exposure to LM-3B11, microvascular and umbilical vein endothelial cells display notable formation of lamellipodial protrusions, membrane structures involved in cell movement and shape changes, in a Src- and PI3K-dependent manner. LM-3B11 induces stronger phosphorylation of Src and Akt compared to other LMs [56]. IHC analyses of non-invasive and invasive breast carcinomas revealed a significant decrease in the number and intensity of α3B-positive vessels compared to normal breast tissue. The LM α3B-chain is often completely absent or minimally detected in blood vessels of invasive carcinomas. In contrast, the α4 chain shows increased expression in non-invasive and invasive carcinomas, particularly in diffusive invasive carcinomas. The α5 chain appears to decrease in expression in some invasive carcinomas, specifically in capillary vessels [56]. This expression pattern might mirror the cytokine profile in the TME, as pro-inflammatory cytokines such as TNF-α downregulate LM-α3B but upregulate the α5 chain in cultured vascular endothelial cells [56, 57]. Therefore, the downmodulation of LM-3B11 in breast cancer could trigger angiogenesis by providing a positive signal for the branching of capillaries infiltrating the tumor.
Several studies have explored how LM-511 influences breast cancer cell behavior, especially regarding invasion and the development of distant metastases. All these studies consistently show that LM-511 actively contributes to the dissemination and metastasis of breast cancer [58, 59]. Although the connection between LM-511 expression and specific breast cancer subtypes is not fully understood, IHC of a limited number of high-grade human breast cancer specimens has demonstrated that the expression of the LM-α5 chain, a component of LM-511, is not exclusive to a particular subtype but is notably elevated in TNBC [58]. These findings suggest the association between high LM-511 expression and the development of aggressive metastatic cancers.
Prostate cancerProstate cancer (PCa) is a prevalent malignancy and one of the most common types of cancer in men. Typically, prostate cancer is an androgen-dependent disease, where androgens bind to and activate androgen receptors in prostate cells, leading to cell proliferation and survival. Evidence suggests that prostate cancers with higher Gleason scores, indicative of more aggressive tumors, are often associated with higher levels of androgen receptor expression [60]. However, some prostate cancers may undergo a transition to a hormone-refractory state, also known as castration-resistant prostate cancer (CRPCa), characterized by increased aggressiveness and resistance to hormonal therapies. As detailed below, various studies have demonstrated an association between LM expression and PCa progression and metastasis. Although differences in LMs between androgen-dependent and CRPCa are not well-established, it is plausible that the heightened aggressiveness of CRPCa might be linked to changes in LM and/or integrin expression patterns in cancer cells [61, 62].
Disassembly of hemidesmosomes, involving proteins such as α6β4 integrins and plectin, is particularly crucial in the initiation of PCa [62]. In normal prostatic tissue or in prostatic intraepithelial neoplastic (PIN) lesions, LM-332 binds to α6β4 integrin to maintain hemidesmosome integrity. However, in PCa, LM-332 forms complexes with α3β1 in actively migrating cells [63]. This switch in integrin receptors is mediated particularly by EGFR signaling, which triggers hemidesmosome disassembly by inducing phosphorylation of the cytoplasmic tail of β4 integrin [64]. Simultaneously, EGFR downregulates LM-332 expression in PCa cells [65]. The downregulation of β4 integrin in aggressive CRPCa cell lines could also be a consequence of hypermethylation of the ITGB4 promoter region or the generation by alternative splicing of a shorter β4 variant that cannot heterodimerize with the α6 integrin chain [66]. In highly metastatic PCa cell lines, there is a coordinated downregulation of β4 integrin and LM-332 chains during the induction of the EMT program by the transcription factor ZEB1 [67]. It is notable that the lack of expression of LM-332 is quite specific for PCa, compared to colon or breast carcinoma samples assayed at the same time [68]. Although protein levels are extremely low or undetectable in PCa tissue, the mRNA for LAMA3, LAMB3, and LAMC2 could be detected in most of the tumor samples by northern blot [65]. This suggests that the loss of LM-332 at the protein level in PCa might be mediated by post-transcriptional mechanisms.
The above results strongly indicate the downregulation of LM-332 and β4 integrin as a central element in inducing an invasive and metastatic phenotype in PCa cells. However, other reports suggest that LM-332 or its subunits might play an active role in promoting PCa motility and invasion. Indeed, while Drake and coworkers showed that ZEB1 repressed LAMC2 and integrin β4 expression in PCa cells, they also found that in vitro migration of PCa cells not expressing LM-332 was enhanced when co-cultured with PCa cells overexpressing LM-332 [67]. This cooperative migration was also observed in organoids formed by PCa cells and fibroblasts, and this enhanced motility of cancer cells in the cocultures correlated with an increased expression of LM-332 chain as well as other ECM proteins [69]. It is interesting that co-culture of fibroblasts and PCa cells also triggered the expression of MMP14 and other proteases, such as cathepsin L, that can induce partial proteolysis of ECM proteins to generate bioactive fragments. In this sense, it has been shown that matriptase digestion of the LM-β3 chain enhances speed migration and directional persistence of PCa cells on LM-332 [70]. Collectively, the results suggest PCa metastasis may be boosted by LM-332 downregulation in PCa cells, which reduce intercellular adhesion, but also by LM-332 upregulation and proteolytic processing in the stromal elements, to trigger PCa motility.
In addition to LM-332, other LMs may induce PCa metastasis in a paracrine manner. In an in vitro study, Graf and colleagues observed that the supernatant of cultured primary human mesenchymal stem cells (MSC) stimulated the migration of highly metastatic PCa cell lines, but not of those with low metastatic potential [71]. The isolation of migratory factors secreted by MSC identified several ECM proteins, including LM-β2- and LM-γ1 chains, as the main proteins with the highest migration-inducing activity. Remarkably, recombinant LM421 induced migration in metastatic PCa cell lines, surpassing the migration response induced by MSC conditioned medium [71]. Additionally, the LM-γ1-derived C16 peptide has been shown to enhance the formation of invadopodia in highly metastatic PCa cell lines [72]. C16 regulates the function of two scaffold proteins important in the formation and function of invadopodia, namely Tks5 (also known as SH3PXD2A or Fish), a SH3-containing protein that interacts with actin-regulatory proteins and signaling molecules, and Tks4 (also known as SH3PXD2B or Tks4/Fish), which plays a central role in the stabilization of MMP14 on the cell surface of invadopodia [72]. It is remarkable that the C16 peptide has also been implicated in the migration of other transformed cell lines.
Ovarian cancerOvarian cancer ranks as the eighth most common cancer among women globally but stands out as one of the most lethal. This heightened lethality is attributed to challenges in diagnosis, often occurring at an advanced stage, and the absence of an appropriate therapeutic regimen [73]. Ovarian cancer is, indeed, a heterogeneous group of diseases, with classification dependent on the cell type undergoing transformation. While the majority (90%) of ovarian cancers originate from ovary epithelial cells, tumors can also develop from germ cells or connective tissue cells (stromal tumors). Epithelial tumors are further subclassified based on histology and characteristics, with the most prevalent being serous ovarian carcinoma (~ 70%), followed by endometrioid (10–20%), clear cell (5–10%), and mucinous (< 5%) carcinomas. The undifferentiated carcinoma subtype is the most aggressive, associated with a very poor prognosis.
Various reports suggest a role for LMs in the pathogenesis of ovarian cancer. Interestingly, the expression and distribution of LMs may vary among ovarian cancer subtypes. LM-332 and LM-511 are frequently detected in serous ovarian cancers, while the BM of mucinous carcinoma also exhibits LM-α4 chain immunoreactivity. In contrast, endometrioid carcinomas show reduced expression of LM-332 but an enhancement of LM-111 [74]. Proteomic characterization of ovarian clear cell carcinomas reveals abundant expression of LM-411 [75], and histological studies indicate immunoreactivity against the LM-γ2 chain [76]. Elevated LM levels have also been detected in the ascitic fluid of patients with peritoneal ovarian cancer dissemination [77].
LMs exhibit both pro- and anti-tumor effects in ovarian cancer. Examination of 370 ovarian cancer samples revealed reduced expression of LAMA3 in carcinoma tissues compared to normal and para-carcinoma tissues [78]. LAMA3 expression was also lower in para-carcinoma compared to normal tissues, indicating a role of oncogenic factors in LAMA3 repression. The mechanism behind this repression may be epigenetic, given that LAMA3 methylation was higher in carcinoma than in normal tissues. Crucially, the analysis of five-year survival rates demonstrated significantly higher recurrence-free survival and overall survival rates in patients with high LAMA3 expression compared to the LAMA3 low-expression group. These findings suggest a clinically relevant role for LM-α3 (and likely LM-332) as a suppressor of ovarian cancer progression.
However, other studies have reported pro-tumoral activities for LM-111 or its peptides in ovarian cancer cells. Intraovarian injection of cancer cells with LM-111 resulted in larger ovarian tumors with higher metastatic potential and ascites production compared to injection with gelatin [73]. Certain LM-111-derived peptides, like AG73 and IKVAV, increased tumor volume, metastatic spread, and ascites production. Notably, the AG73T peptide, a scramble peptide of AG73, caused the disaggregation of ovarian tumor spheroids, rendering these cancer cells more sensitive to cisplatin [73]. Stimulation of ovarian cancer cells with LM-111 or its peptides triggered cell adhesion through β1 integrins, proliferation through the ERK pathway, and survival through the PI3K pathway, along with the upregulation of pro-survival proteins like Bcl2. This provides a mechanistic framework for the pro-tumoral activities of this LM isoform [73]. LM-111, AG73, and IKVAV also induced the concomitant expression of the tumor suppressor p53 and its inhibitor Mdm2, neutralizing the anti-tumoral effect of p53. Interestingly, the LM-111-derived peptides A12 and YIGSR, which reduced tumor growth and metastasis, failed to induce Mdm2 in ovarian cancer cells [73]. However, the mechanism behind the anti-tumoral activity of A12 and YIGSR peptides remains undetermined.
LMs are implicated in the peritoneal metastasis of ovarian cancer through a paracrine mechanism. Overexpression of ETS1, a transcription factor associated with poor prognosis, induced the secretion of large-size exosomes containing high levels of LM-511 in ovarian cancer cells. LM-511 facilitated the uptake of ovarian cancer-derived exosomes by omental macrophages through interaction with the αvβ5 integrin. This uptake polarized peritoneal macrophages toward a pro-tumoral M2 phenotype expressing CXCL5 and CCL2, serving as chemoattractants that direct ovarian cancer cell migration to the peritoneum. Administration of ETS1-expressing exosomes enhanced omental metastasis of ovarian cancer cells, and this effect is inhibited by αvβ5 integrin inhibitors [79].
In ovarian cancer, a noteworthy finding is the presence of a fusion gene known as LAMC2-NR6A1, suggested to act as a driver for oncogenic transformation in ovarian cells [80]. This fusion gene results from the translocation between intron 12 of the LAMC2 gene on chromosome 1 and intron 1 of the NR6A1 gene on chromosome 9. It leads to the production of a short LM-γ2 chain, termed LM-γ2F, which cannot assemble with other LM chains but retains the EGF-like domains of the LM-γ2 DIII region. The LM-γ2F fragment stimulated the EGFR pathway in vitro, demonstrated by the phosphorylation of EGFR, ERK, and AKT, promoting anchorage-independent ovarian cancer cell growth and motility [80]. Knockdown of LM-γ2F reduced, whereas LM γ2F overexpression promoted, the growth of ovarian xenografts implanted in immunodeficient mice. These analyses strongly suggest that the LM-γ2F fusion protein activates important signaling pathways in ovarian cancer progression, although more evidence is needed to confirm its role as a driver mutation for this malignancy.
Oral cancerOral cancer constitutes a substantial portion of cancer cases and deaths worldwide. It encompasses various forms, with squamous cell carcinoma (OSCC) being the most prevalent, affecting tissues like the lips, tongue, hard and soft palate, pharynx, and larynx. Understanding the factors contributing to oral cancer development and dissemination is crucial for effective prevention, early intervention, and improved patient outcomes.
Initial IHC analysis using a LM polyclonal antibody in a limited number of OSCC biopsies revealed a predominantly linear staining pattern at the interface in well-differentiated cases, while poorly-differentiated cases exhibited intense cytoplasmic expression within tumor cells [81]. Recent research has unveiled the multifaceted involvement of LMs in oral carcinogenesis, encompassing interactions with tumor cells, stromal components, and the TME.
LM-332 is one of the most extensively studied LMs in oral cancer, with its expression detected in OSCC but not in dysplastic oral tissue [82]. The LM-γ2 chain is present in tumor-budding areas surrounded by myofibroblasts, indicating its implication in the invasive phenotype of OSCC [83, 84]. These findings propose LM-332, or its γ2 chain, as potential early diagnostic markers for malignant transformation, invasion and poor prognosis. However, in vitro studies present a more intricate scenario, indicating reciprocal regulation between E-cadherin and LM-332 levels. LM-332 binding to α3β1 integrin triggered E-cadherin-mediated adhesion, reducing motility in poorly invasive OSCC cell lines. Silencing of the LM-γ2 chain, which prevented heterotrimeric LM-332 formation, enhanced in vitro migration and in vivo tumorigenicity. LM-γ2 knockdown correlated with reduced α3β3 integrin levels and impaired E-cadherin-mediated cell-to-cell adhesion [84], a hallmark of the EMT program necessary for cancer cell metastasis. Consistently, downregulation of LM-332 expression has been reported upon activation of the EMT program by overexpression of the Snail transcription factor in OSCC cell lines [85].
While the role of heterotrimeric LM-332 remains controversial, there is substantial consensus regarding the association of elevated LM-γ2 chain levels with OSCC cell invasion. The regulation of LM-γ2 chain protein levels involves distinct mechanisms. Long non-coding RNAs CASC9 and BBOX1, typically upregulated in OSCC, act as sponges for miR-545-3p and miR-3940-3p, respectively. This interaction impeded the downregulation of LAMC2 by these miRs, consequently increasing LM-γ2 protein levels [86, 87]. Although EMT induction by Snail overexpression downregulated heterotrimeric LM-332, LAMC2 levels remained unchanged, thereby promoting the deposition of processed LM-γ2 chain, known for its pro-migratory effects [85]. As mentioned below, Snail may also regulate other LM chains, suggesting that a combination of LMs, rather than a single one, is necessary for EMT in OSCC cells. The monomeric LM-γ2 chain potentially affects OSCC cell motility and proliferation by activating the EGFR/ERK signaling pathway [88]. This may occur through indirect mechanisms involving integrin receptors or via direct interaction of LM-γ2 proteolytic fragments derived from domain III with EGFR, as observed in other cancer types [89]. Importantly, positive feedback transcriptional regulation has been proposed for EGFR and LM-γ2 chain in various OSCC types [88, 90]. Monomeric LM-γ2 could also influence OSCC dissemination by affecting stromal cells. Extracellular vesicles enriched in LM-γ2 secreted by OSCC cells stimulates lymphangiogenesis [91], potentially facilitating lymphatic dissemination.
Other LM-332 chains may also impact OSCC carcinogenesis. The amplification of LAMA3 and the increased expression of the LM-β3 chain has been associated with a more aggressive OSCC phenotype, including enhanced invasive potential and metastatic behavior [92]. It has been suggested that the LM-β3 chain enhances the formation and activity of invadopodia [93], specialized structures facilitating ECM degradation and tumor cell invasion. Although the interaction of the LM-β3 chain with integrin receptors is known, the specific LM-β3-induced signals are unknown.
Another LM involved in OSCC carcinogenesis is LM-111. Specifically, the LM-111-derived peptide AG73 positively modulates the migration and invasion of OSCC cells. AG73 triggered OSCC cell invasiveness by inducing syndecan-1- and β1 integrin-signaling pathways that upregulate the secretion of MMP-9 [94], a key enzyme in ECM degradation, angiogenesis induction, and tumor cell migration. The C16 peptide, also derived from LM-γ1 cleavage, promoted OSCC invasiveness by activating the β1 integrin, Src kinase, and the ERK1/2 signaling pathway [95], all of them regulating invadopodia activity, ECM degradation and tumor cell invasion.
The LAMA4 and LAMA5 genes are also implicated in regulating the invasive phenotype of OSCC. EMT effectors, like Snail, upregulate LAMA4 while downregulating LAMA5 expression [96]. Chromatin immunoprecipitation revealed direct binding of Snail to specific promoter regions of both genes. Notably, Snail-induced EMT also alters integrin and non-integrin LM receptor expression; it silences Lu/BCAM, a non-integrin receptor for LM-α5, switches integrin α6β4 to α6β1, and triggers neoexpression of integrin α1β1, a specific receptor for LM-411 [85]. These changes contribute to OSCC invasion by weakening LM-411-mediated adhesion to fibronectin. In addition, EGF and TGFβ1-induced EMT silences both LAMA4 and LAMA5 expression while upregulating LAMC2 [97]. Therefore, different signals may lead to different LM outcomes during EMT in
留言 (0)